| T
H E N I H C A T A L
Y S T |
M
A Y – J U
N E 2001 |
|
Beyond
the unfolding of complete genetic sequences lies the challenge of identifying
and deciphering all the proteins that make up living organisms. Structural genomics—a
new field catapulted into feasibility by the success of gene-sequencing projects
and advances in the tools of structural biology—approaches that task through
the large-scale determination of three-dimensional protein structures.
A protein’s genetic
sequence can provide clues about its function, but a protein’s structure
can better illuminate its biological action and its role in health and disease.
A solved, high-resolution structure maps all the protein’s atoms, exposes
surface topology and inner architecture, reveals electrochemical properties,
and presents a testing ground for possible molecular partners. It paves the
way for advances in structure-based drug design and the development of new medical
devices and materials.
Determining high-resolution
protein structures is often difficult and time-consuming, however. The essential
tools of structural biology—X-ray crystallography and nuclear magnetic
resonance (NMR) spectroscopy—each have their drawbacks. The former requires
crystallization of the proteins, a laborious task, and the latter, though it
uses proteins in solution, is usually slower and is limited to solving the structures
of small and medium-sized molecules.
 |
|
Jacob
Keller, Columbia University
|
|
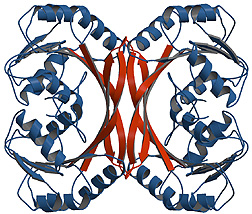
A
protein of Methanobacterium thermoauto-trophicum, MTO146 is one of the
first structures determined by the Northeast Structural Genomics Consortium
and reveals information that could alter the protein’s original functional
assignment. It is also known as precorrin-8W decarboxylase, a label that
Columbia University (New York) crystallographer John Hunt, in whose lab
the structure was solved, expects will change. "Enzymologists categorized
the protein as a decarboxylase indirectly based on its sequence. We’re
not sure if it’s a decarboxylase," he says, "but it’s
unambiguously a methyltransferase" based on the group’s solved
structure of the protein bound to S-adenosylmethionine.
Recounts
Jacob Keller, a research assistant in Hunt's lab, "I was simply playing
around with the structure . . . superimposing structural homologs, when
one of the homologs brought its AdoMet [S-adenosylmethionine] along, fitting
it right into the homologous pocket in MT0146. Since all of the key contacts
were in the right places, I co-crystallized the enzyme with AdoMet, and
the new structure showed AdoMet density half an angstrom from where we
expected it. It was very satisfying."
Although
they still don’t know all the details of the enzyme’s action,
the group’s structural data have convinced them that the protein
adds methyl groups to a vitamin B12 precursor.
Previous work from other groups shows that the enzyme is present in all
organisms that make vitamin B12. As an extra bonus,
it contains a structural motif never seen before: a b-barrel
tetramerization domain.
|
Structural genomics focuses
on cranking out, at industrial speed, thousands of carefully selected structures
from which most others can be predicted computationally with a reasonable degree
of accuracy.
This approach relies on
a belief in nature’s economy—that the countless different proteins
in nature fold into a limited number of shapes and that all natural protein
structures are a subset or combination of these shapes.
 |
|
Andrzej
Joachimiak, Argonne National Laboratory
|
|
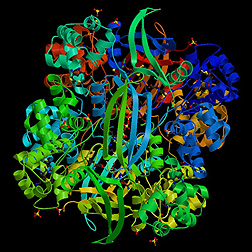
Originally
thought to look like a DNA-binding protein, this protein structure solved
by Andrzej Joachimiak of Argonne (Ill.) National Laboratory, who leads
the Midwest Center for Structural Genomics, turned out to be an enzyme
with cyanase activity. The work illustrates how structural genomics can
shed light on the evolution of protein function. This cyanase converts
toxic isocyanide to ammonia and carbon dioxide, making it potentially
useful as a detoxifying agent. It is a decamer composed of five dimers.
The enzyme’s amino acid sequence is not similar to any other known
protein, and apparently neither is its structure. "The subunits of
cyanase are arranged in a novel manner both at the dimer and decamer level,"
according to Joachimiak.
|
The key to structural
genomics is to group proteins into families of similar structures based on their
sequences. Then, based on the known structure of at least one protein in a family
and using a computational technique called homology modeling, a good guess can
be made about the shapes of other proteins in the family. Estimates of the number
of protein structure families range from 30,000 to 50,000—orders of magnitude
smaller than the total number of proteins in nature.
Thinking
Globally, Acting
Locally
Currently, there is funding
for structural genomics projects in the U.S., the European Union, Japan, China,
Canada, and Israel. (In early April, representatives from four continents gathered
in Virginia to discuss goals, progress, and policy issues; see "International
Airing.")
Pharmaceutical companies
and biotech start-ups are also committing to structural genomics, primarily
to aid drug discovery.
The publicly funded U.S.
effort is spearheaded by NIGMS, which
last September launched the Protein
Structure Initiative and will spend $150 million over the next five years
on seven structural genomics pilot research centers, including one co-funded
by NIAID (see "The
First Seven."). NIGMS expects to fund a few additional centers this
September.
These pilot centers will
develop new techniques to streamline and accelerate every step in structural
genomics, from choosing which protein structures to solve to cloning and purifying
the proteins, determining the structures, and depositing the data into the Protein
Data Bank (PDB), an online database of macromolecular structures, maintained
by the Research Collaboratory for Structural Bioinformatics.*
In five years, each of
the centers will ramp up to a production level of 100 to 200 structures annually
at a significantly reduced cost per structure. Using traditional techniques,
it takes weeks to months—and an average of more than $100,000—to solve
the structure of a single globular, soluble protein. More recalcitrant proteins,
such as membrane proteins, are even more challenging.
One long-term goal of
the NIGMS project is to develop a public library of nature’s protein shapes
that integrates sequence, structural, and functional information. This library
should enable researchers to use genetic sequences to predict the approximate
structures—and possibly the function—of any protein.
To build this public resource,
NIGMS is enlisting its pilot centers to determine the structures of one or two
representative proteins from each of thousands of different structural families.
Ten thousand unique protein structures should be solved over 10 years, which
includes the current five-year scale-up phase, then five more years at full
speed.
Currently, of the 15,000
structures that have been deposited in the PDB,
less than 4,000 are of unique proteins, defined as those whose sequences are
less than 90 percent identical. And the solved PDB structures represent only
about 1,500 families. By determining 10,000 protein structures from almost as
many families, the Protein
Structure Initiative would more than triple the number of unique structures
available and would provide more thorough coverage of structural families.
One catch at this early
stage is that there are many different ways to group proteins into families.
The five-year pilot period should provide time to determine whether any particular
method is better than the others.
The project also seeks
to identify new folds. Proteins with the same fold have similar overall shapes
but no detectable sequence similarity. Such proteins have the same types of
structural components connected in the same order. Studying folds could reveal
the physical and chemical principles that determine how proteins form their
three-dimensional structures.
Scientists estimate there
are only a few thousand folds–considerably fewer than the number of structure
families—and only 700 of these are represented in the PDB.
Just
Data Gathering?
In its early days, structural
genomics was criticized by those who believed it was a rote exercise devoid
of the creativity and intellectual challenge that characterize high-quality
scientific research. Although such concerns are less common now, says John
Norvell, who directs the NIGMS initiative, "it’s certainly true
that structural genomics isn’t hypothesis-driven. It’s discovery-driven"—much
like Darwin’s detailed observations and descriptions of finches, barnacles,
and other creatures, which led to his theory of evolution, Norvell observes.
Although it is clearly
too early to predict the eventual impact of the Protein
Structure Initiative, like its predecessor, the Human
Genome Project, it promises to open a whole new chapter in biomedical research.

*
The Research Collaboratory for Structural Bioinformatics
is a joint project of Rutgers University,
Piscataway, N.J., the San Diego Supercomputer
Center at the University of California at San Diego, and the National
Institute of Standards and Technology, Gaithersburg, Md. It is supported
by funds from the National Science Foundation.
|
International
Airing
For
three days in early April, Airlie Conference
Center, a restored estate in the Warrenton,Va., countryside, sounded
like a miniature United Nations—but with a scientific twist. The
voices speaking to each other in French, German, English, Italian, Chinese,
and Japanese were those of participants in the Second
International Structural Genomics meeting. They discussed policy issues,
bottlenecks, and the status of their structural genomics projects. The
Airlie Agreement, which is available online,
presents the consensus of the group on various policy issues.
Many of the discussions
focused on balancing two different goals—timely release of all structural
genomics data to the public and respect for intellectual property laws
that vary significantly in different countries. The group was particularly
concerned about the possibility that patents could be based solely on
the submission of three-dimensional structural coordinates, without any
identified nontrivial utility.
The participants
agreed that for projects with public funding, researchers must deposit
atomic coordinates and associated experimental data into the Protein
Data Bank immediately after their determination and release most of
these to the public soon thereafter. In some cases, the researchers may
delay data release for up to six months to facilitate patent filing.
They also agreed
that, although the goal of the field is to maximize efficiency, obtaining
high-quality structures is of primary importance. Projects must not compromise
quality for speed. Nor, however, should data release be "unduly delayed"
while researchers endlessly refine their structures. They declined, however,
to specify numerical criteria for when a structure is considered complete
and ready to deposit.
–Alisa
Zapp Machalek
|
|
The
First Seven
The
NIGMS Protein Structure
Initiative is currently supporting projects at seven research
centers to determine thousands of protein structures; study the relationship
between genes, protein structure, and protein function; and develop new
techniques. NIGMS will spend more than $150 million on these projects
over five years, making it the world’s single largest supporter of
structural genomics. The centers, each a collaboration among multiple
institutions, are:
Berkeley Structural Genomics Center.
Will focus on two closely related bacteria with extremely small genomes—Mycoplasma
genitalium and Mycoplasma pneumoniae—to study proteins
essential for independent life. Aims to accelerate structure determination
by X-ray crystallography.
The Joint Center for Structural Genomics.
Will initially focus on novel structures from the roundworm Caenorhabditis
elegans and on human proteins thought to be involved in cell signaling
and will determine the structures of similar proteins from other organisms
to ensure the inclusion of the greatest number of different protein folds.
Aims to develop high-throughput methods for protein production, crystallization,
and structure determination.
The Midwest Center for Structural Genomics.
Will select protein targets from the domains Eukarya, Archaea, and Bacteria,
with an emphasis on previously unknown folds and on proteins from disease-causing
organisms. Aims to reduce the average cost of a protein structure from
$100,000 to $20,000.
New York Structural Genomics Research
Consortium. Aims to develop techniques to streamline every step
of structural genomics and to solve several hundred protein structures
from humans and model organisms.
Northeast Structural Genomics Consortium
. Using both X-ray crystallography and NMR spectroscopy, will target
proteins from various model organisms—including the fruit fly, yeast,
and the roundworm—and related human proteins.
The Southeast Collaboratory for Structural
Genomics. Emphasizes technology development, especially for automated
crystallography and NMR techniques. Will analyze part of the human genome
and the entire genomes of two model organisms genetically and biochemically
similar to humans—the roundworm Caenorhabditis elegans and
the high-temperature microbe Pyrococcus furiosus.
TB Structural Genomics Consortium).
A collaboration of scientists in six countries formed to determine and
analyze the structures of about 400 proteins from Mycobacterium tuberculosis.
Will optimize the technical and managerial underpinnings of high-throughput
structure determination and will develop a database of structures and
functions. NIAID, which is cofunding
this project, anticipates this information will lead to new and improved
drugs and vaccines for tuberculosis.
More information
about the NIGMS Protein Structure Initiative is available at its web
site.
|
Return to Table of Contents