| T H E N I H C A T A L Y S T | N O V E M B E R – D E C E M B E R 2000 |
|
|
|
NIH Research FestivalRUNNING WITH THE GENOME:NIH GETS TO WORK WITH THE'WORKING DRAFT' |
|
The "working draft" sequence of the human genome, delivered to the desktops of the biomedical community this summer, may become the most "dog-eared" work-in-progress the world has ever known.
Unlike the first draft of a medical text that requires corrections and updates before it is finally printed, the genome’s first draft is out there, and some NIH investigators are using it to make new discoveries—while others are arranging the data to make the material even more meaningful as a resource to the research community.
Simultaneously, the working draft is constantly growing, as sequencing data pours in, nonstop, from centers around the world. The working draft—which was 85–90 percent complete when it was announced in June—should be entirely filled in by year’s end. The finished product (see description below) is anticipated in the year 2003, Eric Green said at the NIH Research Festival plenary session on "The Utility of Whole Genome Sequences: Early Glimpses of the Sequence-Based Era."
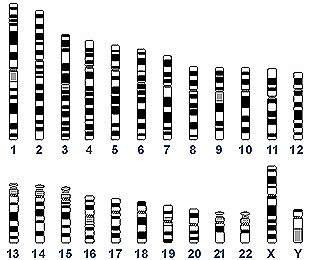
Calling the human genome a "24-volume encyclopedia set—volumes 1–22, X, and Y," Green, chief of NHGRI’s Genome Technology Branch and director of the NIH Intramural Sequencing Center, presented an overview of the fundamentals of the Human Genome Project (HGP).
Rounding out the session, NCBI’s Greg Schuler described the computational programs rapidly being developed to tackle the task of assimilating and utilizing the data—and Nick Ryba, chief of the NIDCR Taste and Smell Unit, reported on how strategic use of the HGP’s data aided the discovery of a new family of receptors (see "An Acquired Taste").
 |
|
Eric
Green
|
The ABCs of Sequencing
Stage 1 of the HGP involved studying and organizing the DNA from each chromosome in a process known as mapping. For physical mapping, each chromosome is broken up into larger or smaller pieces and isolated as DNA clones. These clones are characterized and overlapped on one another by identifying regions in common. The resulting "contigs" constitute a contiguous segment of the starting DNA. "Like a jigsaw puzzle, the larger clones are at first easier to put together into contigs than the smaller clones," said Green.
Two DNA cloning systems have been instrumental to the HGP’s efforts to map the human genome. In one, the larger pieces of cloned DNA are isolated as yeast artificial chromosomes (YACs); in the other, smaller pieces of cloned DNA are isolated as bacterial artificial chromosomes (BACs). "Each YAC provides multiple pages from one of the hypothetical chromosomal volumes, or, roughly, chapter-size pieces of cloned DNA," Green explained; each BAC clone is approximately page-size. Because of larger size and availability, YACs play a dominant role in constructing the first-generation physical maps of the human genome.
Stage 2 of the HGP, Green continued, involves sequencing the organized DNA—using what is termed a "sequence-ready contig map," or a series of overlapping BACs that map to a certain region of a human chromosome–to construct each human chromosome page by page.
The fundamentals of DNA sequencing—developed in 1977 by Fred Sanger, a British scientist who received his second Nobel Prize in chemistry for work in this field—have not changed, but sequencing efficiency has—dramatically. In one year, Green said, a single person can produce more than one million bases of sequence. "Shotgun sequencing" is the most commonly used method: "BAC clones are selected and large amounts of DNA made (like taking a page and making lots of xerox copies), then randomly fragmented (putting these pages in a paper shredder)." Thousands of these fragments are sequenced and assembled by a computer program. In this way, each fragment of the human genome is essentially sequenced multiple times. Checking for redundancy among these multiple reads ultimately produces a highly accurate "working draft" sequence.
"Sequence finishing" is a term used to describe the hard polishing of the sequence—getting additional sequence reads to improve accuracy and cover gaps—refinements that yield a highly accurate sequence, thereafter known as a "finished" sequence.
|
According to Francis Collins, NHGRI director and head of the Human Genome Project, the public working draft version of the human sequence can be found in its most useable forms at the following web sites:
|
Accomplishments of the HGP to date include complete sequencing of a number of microbial genomes, in addition to the first eukaryotic organism, the common brewer’s yeast reported in 1997, and the first multicellular genome, the nematode Caenorhabditis elegans, in 1998. Earlier this year, a collaboration between Celera Genomics, of Rockville, Md., and the HGP resulted in the completion of the sequence for the fruit fly, Drosophila melanogaster.
The G5
The complete working draft of the human genome is expected by year’s end, with a projected completion of the finished sequence in the year 2003. The working draft of the mouse genome should be available early next year and the finished sequence by 2005, Green said. To meet the deadlines for the human genome sequence, five major sequencing centers, collectively known as "the G5," were responsible for sequencing approximately 85 percent of the human genome. About 12 other centers were responsible for sequencing the rest.
The G5 are:
![]() Washington University Genome Sequencing
Center, St. Louis
Washington University Genome Sequencing
Center, St. Louis
![]() Whitehead Institute for Biomedical Research/MIT Center
for Genome Research, Cambridge, Mass.
Whitehead Institute for Biomedical Research/MIT Center
for Genome Research, Cambridge, Mass.
![]() Baylor College of Medicine, Human Genome
Sequencing Center, Houston
Baylor College of Medicine, Human Genome
Sequencing Center, Houston
![]() U.S Deparment of Energy Joint Genome Institute, Walnut Creek, Calif.
U.S Deparment of Energy Joint Genome Institute, Walnut Creek, Calif.
![]() The Sanger Centre, Hinxton, United Kingdom
The Sanger Centre, Hinxton, United Kingdom
The entire human genome of about three billion bases will be able to fit, roughly, on one CD-ROM, Green said. With such rapid accumulation of bulk data, the next phase of the HGP will concentrate on analysis and interpretation and developing the software tools to expedite these efforts. He expects the next few decades to be spent seeking better ways to identify genes and key sequences that determine how, when, and where genes are turned on.
 |
|
Greg
Schuler
|
The year 2000, Green said, has been the turning point.
It’s a Blast!
"One of the great things about the Genome Project," said Greg Schuler, NCBI staff scientist, "is that you don’t have to wait until it’s finished to start making use of the data."
Schuler and his NCBI colleagues are getting creative with the sequence data generated by the HGP—data that are regularly downloaded within 24 hours from sequencing centers into the public database, GenBank. Schuler and company are working to make the system user-friendly and optimized for the needs of researchers.
GenBank is equipped with several features that provide pertinent information on the quality of particular working draft sequences, such as redundancy, fragment number, and base quality scores. It also allows users to look at different types of sequence entries–for instance, by chromosomes or large contigs. In many cases, unique qualifiers are used to label data so that known genes and their protein products are distinguished from predicted genes and their protein products. The standard tool for sequence analysis, BLAST (Basic Local Alignment Search Tool), offers multiple searching options: "For example," Schuler said, "suppose you had a query protein and its gene sequence. You could use the software tBLASTn to scan the draft of the genome for any matches in humans or other organisms." Alternatively, he added, one could use the genome to come up with predicted proteins and make a searchable database followed by a BLASTp search and compare results with the query protein. With a predicted protein sequence, one could search for common protein domains to get information about function or look at the percentage of conserved domains.
Schuler emphasizes that
the genome sequence provides a uniform frame of reference (or coordinate sytem)
for relating the positions of disparate types of genomic features, such as genes,
mapped markers, and common sequence variations. The team has also developed
a 'locus identifier' and a program called 'Refseq,'
which aims to establish one sequence entry for each naturally occurring DNA,
RNA, and protein molecule. ![]()
In collaboration with HHMI investigator Charles Zuker and his group at the University of California, San Diego (UCSD), we have used the "working draft" to help uncover a family of taste receptors. Aversive reactions to bitter tastes protects animals from ingesting toxic compounds. But until human genome sequence information helped identify a new family of receptors we had little idea how many chemically unrelated compounds could all taste bitter. G-protein coupled receptors (GPCRs) have been implicated in mediating bitter taste, and last year, the ability of humans to taste a specific bitter substance was genetically linked to a region of chromosome 5 (5p15). The NIDCR-UCSD group used tools such as open reading frame-finder coupled with BLASTp and hydrophobicity analysis to identify a new candidate GPCR (T2R-1) in sequence from this interval. But what was really exciting was that tBLASTn searches of draft sequence indicated that T2R-1 was a member of a large family of GPCRs clustered in just a few regions of the genome, all implicated in controlling bitter taste in mammals. Using molecular biology techniques, we cloned homologous receptor genes from rodents and examined where T2Rs are expressed. As expected, for sensory receptors, T2Rs were found in subsets of taste receptor cells. Then, using a cell-based assay system, we showed that at least some T2Rs responded to bitter-tasting compounds. For example, the highly toxic and bitter molecule cycloheximide selectively activated mouse T2R-5. Intriguingly, the ability of mice to taste low concentrations of cycloheximide has been mapped to a locus at the distal end of chromosome 6, just where the T2R-5 gene is also found. Moreover, nontaster mice have five mutations in T2R-5, and these affect the sensitivity of the receptor in the cellular assay, strongly pointing to a genetic explanation for a behavioral trait. Why do many unrelated toxic compounds taste bitter? It turns out that taste cells containing one T2R also express most of the other T2R genes. The brain interprets information about patterns of cellular activation rather than which receptor is involved. This means that substances activating different T2Rs are likely to taste similar. There is already
considerable interest in exploiting this information to modify taste.
For example, certain drugs for AIDS, heart disease, and depression taste
so bad or so ruin the flavors of food that patients abandon life-saving
medications. But the main focus of the NIDCR group will be trying to extend
our knowledge of how taste works, ultimately to understand how information
is encoded in the tongue, transmitted to the brain, and decoded there
to produce the familiar sensations that contribute so much to our everyday
enjoyment. For further information
about T2Rs, see "A
Novel Family of Mammalian Taste Receptors" and "T2Rs
Function as Bitter Taste Receptors," printed in the March 2000
issue of Cell. |
|
GenBank Field Guide, Dec. 14–15 For a free lecture and workshop on GenBank and related NCBI molecular biology databases, register online. Address additional questions to Peter Cooper. |
