H O T M E T H O D S C L I N I C
by Chetan Seshandri, Mary Huckabee, Nicole Simone, Michael Emmert-Buck,
and Lance Liotta, Laboratory of Pathology, DBS, NCI,and Robert Bonner,
Laboratory of Integrative and Medical Biophysics, NICHD
LASER
CAPTURE
MICRODISSECTION
FACILITY
BRINGS
MOLECULAR
PATHOLOGY TO
NIH COMMUNITY
| Disclaimer: | Mention of specific products in this article does not constitute an endorsement of those products, nor does it signify that other similar products are less desirable. |
| P |
Over the past decade, the polymerase chain reaction (PCR) has allowed the robust analysis of very small amounts of tissue; nevertheless, a major obstacle in applying PCR to tissue is the inherent heterogeneity of the cell populations. In a gross biopsy specimen from a cancer patient, for example, the neoplasia may only constitute a small fraction of the total tissue mass. Inflammatory cells, stromal elements such as fibroblasts, and normal tissue will inevitably be present and will mar the molecular analysis of the tumors. The tumors themselves may be polyclonal in origin, further complicating genetic analysis. Finally, haphazard sampling of the cells in the tissue may disturb the normal gene expression pattern regulated by cell-cell and cell-matrix interactions and other local environmental factors.
To address these issues, microdissection techniques have been developed, and these have moved from comparatively tedious and difficult manual maneuvers to the more elegant approach used by Shibata (2). In this technique, a protective dye is applied to the areas of interest and ultraviolet radiation is used to ablate the unprotected tissue. The tissue areas of interest remain untreated and can be scraped manually and collected for study. A more automated technique involves placing the tissue section on a film and using an ultraviolet laser to ablate the unwanted regions, leaving islands of exposed regions of interest (3). The selected islands are then procured with a needle. But even this method becomes tedious when small foci are the targets of dissectionthe desired areas must still be circumscribed by the ablation and collected one by one.
The newest generation of automated microdissection is laser capture microdissection (LCM) (4),developed via the intramural collaborative efforts of NCI and BEIP. The technology is being commercialized through a Cooperative Research and Development Agreement with Arcturus Engineering, Inc.
Unlike previous microdissection methods, LCM operates by positive rather than negative selection. First, a clear-transfer film is applied to the surface of the tissue. An infrared laser melts the film in very focused areas, targeting for capture only the cell(s) of interest.
![[ LCM Schematic ]](images/lanceschema.gif) |
The commercially available LCM system, called PixCell, offers additional advantages: The transfer film is on the underside of a cap that fits into a standard 500µl Eppendorf tube, which facilitates digestion of the tissue and procurement of a PCR template. Also, a soon-to-be automated process of loading and unloading the caps will greatly reduce the possibility of contamination, an important consideration for PCR.
NCI is making its core LCM facility available to all NIH investigators. Several LCM microscopes will be available, with technical assistance and pathologists standing by. Interested investigators can call and set up an appointment to bring their material. If the starting material is properly prepared, the microdissection session can be quite short, and the investigator can leave with the microdissected cells ready for analysis. LCM training conferences are being held every two months. To register for the course, contact Robert Bonner at 301-435-1946. You must attend the LCM training conference before using the Center.
Embedding
Typically, tissues must be fixed, dehydrated, cleared, embedded, sectioned, and mounted. Each of these steps can influence the efficiency of the LCM transfer and the subsequent PCR analysis. We have found the following protocol works best for LCM of paraffin-embedded tissues (PET).
Tissue Preparation
1. Fix tissue in 10% neutral buffered formalin (NBF) or 70% ethanol for 2.5-4.0 hours for 3-mm-thick tissue slices.
Fixation is performed to preserve the morphology of the living tissue, but it does not necessarily have a beneficial effect on the DNA. Formalin, one of the most popular fixatives, cross-links DNA to protein, thus making the DNA molecule rigid and susceptible to mechanical shearing during the handling of DNA-containing aqueous solutions (5). Direct cross-linking of DNA strands or of DNA to protein will also prevent polymerase reading through these sites and reduce the effective DNA length for PCR. The DNA yield from formalin-fixed tissues decreases with prolonged fixation time, but the loss is acceptable if routinely processed specimens are fixed for less than 24 hours. DNA yield and quality are improved following ethanol fixation and appear unaffected by the duration of fixation (6). We are now developing ethanol-based fixation procedures.
2. After fixation, the following steps are performed in an automated tissue-processing machine.
Processing can be completed routinely overnight, or it may be handled on an accelerated basis. We have found no change in the LCM transfer efficiency for tissues processed via the accelerated cycle.
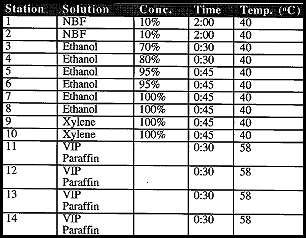 |
3. Block the wax and specimen out in a mold.
4. Cut sections on a clean microtome with a clean blade.
5. Float paraffin ribbons on 43-44oC deionized water (NO ADHESIVES).
6. Mount on plain, uncoated glass slides.
7. Dry slides in a 37oC oven for one hour.
Paraffin embedding can be used for samples destined for DNA analysis, but it causes damage to RNA; thus, frozen tissue should be used for RNA analysis. Embedding and sectioning frozen tissue, as described by Kiernan (7), yields slides suitable for LCM .
Staining
Staining should be performed as indicated below, using solution baths that are replaced regularly. Note that the times for hematoxylin and eosin staining are dramatically shorter than normal histology would require. We believe longer staining will reduce the efficiency of transfer and decrease the length of DNA fragments that can be recovered. For DNA analysis of a paraffin-embedded section, begin staining procedures with deparaffinizing (step #1 below). For a frozen embedded section for RNA analysis, begin with step #6, using 70% ethanol to quickly fix the section.
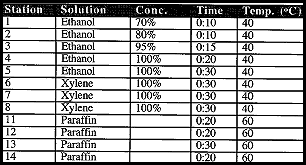 |
1. Soak slides for 5 min in xylene to deparaffinize them.
2. Rinse for an additional 5 min in clean xylene.
3. Dip slides 12 times (approx. 5 sec each dip) in 100% ethanol.
4. Dip for 5 seconds in a clean bath of 100% ethanol.
5. Dip slides 12 times (approx. 5 sec each dip) in 95% ethanol.
6. Dip slides 12 times (approx. 5 sec each dip) in 70% ethanol.
7. Dip slides 12 times (approx. 5 sec each dip) in purified water.
8. Dip the slides for 10-15 sec in Mayer's hematoxylin.
9. Dip slides 12 times (approx. 5 sec each dip) in purified water.
10. Submerse slides for 10-15 sec in bluing reagent.
11. Dip slides 12 times (approx. 5 sec each dip) in 70% ethanol.
12. Dip slides 12 times in 95% ethanol
13. Submerse slides in Eosin Y for 30-60 sec.
14. Dip slides 12 times (approx. 5 sec each dip) in 95% ethanol.
15. Dip slides 12 times in a clean bath of 95% ethanol.
16. Dip slides 12 times in 100% ethanol.
17. Submerse slides for 30-60 sec in xylene.
18. Shake off excess xylene and wipe slides carefully with particle-free paper towel or tissue
19. Air dry slides at least 2 min to allow xylene to evaporate completely.
Microdissection (LCM)
Slides with fixed, sectioned, stained tissue should be carefully labelled, including information on sample thickness, staining, and coating of slide, as well as identification of the tissue targeted for transfer (e.g., invasive cancer, normal epithelium, lymphocytes), fixation protocol, and the actual section number to be captured if it is a serial section.
The sample thickness will be useful in automatic computation of the volume of transfer from spot size, number of laser spots, and the percent transferred within a spot. Prepared slides are then brought to the LCM Center, located in Building 10, Room 2C-401.
To sign up for a user slot, call Bob Bonner at 435-1946, Lance Liotta at 496-2035, or Michael Emmert-Buck at 496-2912. Collaborators may set up as much as a three-hour time block to work with a Laser Lab staff member. These times are either 10 a.m.-1 p.m. or 2 p.m.-5 p.m. daily.
At the Center, a computer program created by DCRT will guide users in turning on the microscope, microscope monitor, vacuum pump, and the laser, and will establish a file folder for each user's study.
The computer will prompt the user to enter data on the slide number and the specific tissue targeted for transfer. Careful recording of this informatin will allow correlation of LCM images relative to the coverslipped slide, permitting use of adjacent sections for high quality roadmap images.
After entering additional cap and identification information for tracking the job, the user can add additional optional information. Next, the program prompts input of the laser parameters, including spot size, pulse power, pulse duration, and sample thickness. These critical values are used by the computer to control the laser. Presently, spot size is set by the user.
A 60-µm beam is selected by using a weight with a defocusing lens, and a 30-µm beam is obtained by using a weight without the lens. This value is important to include for reference.
We recommend setting the pulse power to 30 mW, the pulse duration to 50 ms, and the beam size to 60 µm. A 60-µm beam should be used for homogeneous tissue for more rapid transfers. When the targeted cell groups within the tissue are smaller than 60 µm, then the 30-µm beam will be used.
After valid entries have been recorded for user, study, slide number, cap information, laser pulse duration, and pulse power, LCM will proceed, with all this information recorded in the data record file of the transfer, along with any images taken.
The user can acquire images and begin LCM transfer. The estimated percent transferred can be adjusted after the dissection if the user feels that the default 90% value is incorrect.
Next, the user selects a coverslipped slide and obtains roadmap images. If the user has serial sections, it is best to coverslip the middle slide. The computer allows users to select, annotate, display, and review roadmap images, with options for voice annotation and web capture of these images.
For all subsequent procedures, users should wear gloves to avoid contaminating the microtransfers or leaving finger oils on extraction vessel tubes. Finger oils make it difficult to write on the tubes.
During the transfer process, users center the viewing area with a joystick and then rotate the microscope turret to center the 4X objective over the stage. At this magnification, the view in the eyepiece is almost exactly the size of the film surface on the cap. This is the region within which transfers can be made on a single cap. Users position the slide to cover vacuum holes and then use the cap-loading device to load a cap onto the cap rack, taking care to avoid any contact with the film surface. Multiple caps can be loaded and used successively.
Next, users operate the cap-manipulating arm to position and drop the cap onto the tissue. The position of the laser spot is indicated by the low-intensity aiming beam on the computer screen. Tissue can be selected and transferred at 4X, 10X, 20X, or 40X magnification.
Users next fire a series of laser pulses, moving the stage between pulses to select different target clusters. Before the tissue is removed, users may take and annotate "before dissection" pictures. They then quickly remove the cap. Time is of the essence here to prevent the polymer from deforming and contracting without removing all the tissue.
Users check the original tissue sample to verify the transfer has occurred and may take and annotate "after dissection" pictures before positioning a clean glass slide on the center of the microscope stage. Users may also photograph the tissue that has been removed by snapping a "cap image."
Before concluding, users review their images, edit any parameters that have been altered, write all the data to their file, label the Eppendorf tube that will hold the sample (date, author, study, case number, slide number, tissue type, number of cells, etc.), verify that it contains sufficient digestion buffer (e.g., 50 µl buffer aliquoted from solution containing 20 µl 1% proteinase K, 480 µl 10mM Tris-HCl [pH=8.0], 1mM EDTA, 1% Tween-20), cap the tube, and turn it upside down, shaking gently until the buffer drops to the cap surface.
Molecular Analysis
Users then return to their labs with the LCM samples and conduct molecular analyses. Thanks to the greater homogeneity of populations of cells extracted via LCM, molecular analysis is significantly enhanced and can be extended to more applications. DNA and RNA extraction and amplification has become more efficient; cDNA libraries (8) and microarrays (9) can be used to discover the functions and interactions of genes.
For DNA extraction, procedures used by Emmert-Buck (10,11) are recommended. Buffered samples from the LCM are incubated overnight at 37° C and are centrifuged for 5 min. Next, the tube caps are removed and the samples boiled for 8 min at 95° C to inactivate proteinase K. The samples can then be used as templates for PCR.
For RNA isolation, procedures modified from the Stratagene Micro Isolation Kit, Catalog #200344, may be used and scaled down for size and purity according to the protocol in Schena et al. (9).
Troubleshooting Tips
Tissue Prep
1. Guard against excessive adhesion of tissue to the slide. For a successful LCM transfer, the strength of the bond between polymer film and the targeted tissue must be stronger than that between the tissue and the underlying glass slide. We observe reduced efficiency of transfer with slides that have been charge-treated (poly-L-lysine) to increase tissue adherence to the slide. Baking the sample onto the slide may denature the interface surface and bond it too strongly. Some histopathology labs use an adhesive in the water bath to improve the tissue section adhesion to the slide, and this may result in reduced or variable LCM transfer. However, when we use plain glass slides that are not charged or coated, we achieve consistent (100 out of 100 30-60µm diameter spots) LCM transfers from 4-10µm thick PET sections.
2. Mount the tissue as close to the center of the slide as possible; otherwise it may be difficult to lock the slide down to the stage and dissect the area of interest.
3. The microtome used to cut sections should be kept clean: excess paraffin and tissue fragments should be wiped away with xylene and a fresh microtome blade should be used for each block.
4. Store prepared tissue slides at a moderate temperature (18°C) and humidity (45%) until required for LCM transfer. Stain tissue just before LCM transfer time.
Staining
1. Filter hematoxylin and bluing solution to remove precipitates; do not allow 100% ethanol to hydrate; be sure to completely deparaffinize slides with xylene. After staining, a final xylene rinse is most critical for tissue transfer. Whenever transfer is inadequate, you can repeat staining procedure steps 11, 12, 14-19.
2. Sections must be dehydrated and not coverslipped for effective LCM transfer. This makes the staining appear darker and more granular. Where the polymer melts and bonds to the targeted tissue, it will appear lighter. A diffuser film on the slides, which we can supply, will dramatically improve imaging.
Transfer
If nonspecific transfer is noted in the "CAP IMAGE," it can be removed. A methanol wash of the cap surface will eliminate loose dirt and tissue. For material that is more firmly held, apply a piece of Scotch tape to the cap surface and peel it away quickly. The removed debris can be visualized by placing the piece of tape on a clean glass slide.
Extraction and Amplification
1. The cap should be inserted into the standard Eppendorf 0.5-ml tube with a special tool to ensure there is a 0.0625-in gap between the top of the cap and the tube rim..
2. There is a considerable range in the nucleotide sequence length that can be expected to produce good PCR results. Some investigators estimate the limit on amplification products from PET sections to be as low as 80-170 base pairs (12). However, most of the current literature (12-15) and our own experience suggest that products around and below 400bp can be expected. We have succeeded in amplifying a 220-bp product from a cap with as few as ten 60µm transfers containing 100 cells. The template concentration was 2 cells/µl.
3. For fragments less than 250 bp, or analysis requiring resolution of fragments of similar size, separation of the PCR products by polyacrylamide gel electrophoresis (PAGE) is recommended. With the small amounts of tissue garnered from microdissection, PCR products can be labeled with 32P for increased sensitivity (10,11).
Concluding Remarks
The protocol presented here has been verified by a user group trial and represents a guideline for the present use of the technology. But the technique is evolving rapidly. Modified protocols are being developed for the use of alternative staining techniques, including immunohistochemistry.
Better imaging during transfer, for example through an index-matching fluid, is also being explored. Although dissection is recommended at 30 or 60µm for reproducibility, preliminary results indicate that transfers as small as 10µm may soon become standard.
LCM will, for the first time, allow researchers to study any disease process at the molecular level. In oncology, this will lead to an understanding of the progression of cancer through various premalignant stages, all of which can be dissected and analyzed. In infectious diseases, it will lead to a more precise understanding of infection, as well as the effects of treatment, at the cellular level. In developmental anatomy, we can begin to catalog the myriad focal changes that have far-reaching effects. We expect that as the technology itself evolves, laser capture microdissection will continue to redefine the state of the art in these fields.
Resource People
RNA recovery: Kristina Cole, NCI, 6-3379
DNA recovery: Jeff Lee, NCI, 6-2912
Human tumor LOH: Michael Birrer, NCI, birrerm@bprb.nci.nih.gov
Human pathology: Rodrigo Chuaqui, NCI, 6-3379
cDNA libraries: David Krizman, NCI, 5-5155
LCM commercialization: Arcturus Engineering, Inc. (408 654-7937)
The LCM web site, which includes a description of the core lab, procedures, instruments, and protocols is at <http://dir.nichd.nih.gov/lcm/lcm.htm>
|
CATALYTIC REACTIONS Below are comments we received in response to questions posed or issues raised in recent issues. On collaboration quandaries In my opinion, there are four major problems with collaborations. First is the requirement that all authors be fully responsible for all data included in a publication. A tremendous amount of trust is required, especially with a long distance collaboration or a collaboration between persons in very different fields. The second is that genrally first or senior authorships carry the most weight in hiring, promotion, or tenure decisions since, on average, only one-half of collaborative papers will result in a senior or first authorship. The third is that for a tenure-track person, collaboration usually means working with a more established lab. Even if a tenure-track person is senior author on a paper with a better known researcher, the paper may often be associated with the "bigger" name in oral presentations by others in the field. The fourth is that letters of recommendation for promotion and tenure from collaborators usually carry less weight. One often has to balance the value of collaborating with a giant in one's field against losing that person as a recommender. -- anonymous |
2. D. Shibata, D. Hawes, Z. Li, et al. "Specific genetic analysis of microscopic tissue after selective ultraviolet radiation fractionation and the polymerase chain reaction." Am. J. Pathol. 141:539-543 (1992).
3. M. Bohm, I. Weiland, K. Schutze, et al. "Microbeam MOMeNT: noncontact laser microdissection of membrane-mounted native tissue." Am. J. Pathol.. (in press).
4. M.R. Emmert-Buck, R.F. Bonner, et al. "Laser capture microdissection." Science 274:998-1001 (1996).
5. C. Mies. "Molecular biological analysis of paraffin-embedded tissues." Human Path. 25:555-560 (1994).
6. D. Crisan and J.C. Mattson. "Retrospective DNA analysis using fixed tissue specimens." DNA and Cell Bio. 12:455464 (1993).
7. J.A. Kiernan. Histological and Histochemical Methods: Theory and Practice. Second Edition. Pergamon Press, New York. (1990).
8. D.B. Krizman, R.F. Chuaqui, P.S. Mettler, J. M. Trent, P.H. Duray, W.M. Linehan, L.A. Liotta, and M.R. Emmert-Buck. "Construction of a representative cDNA library from prostatic intraepithelial neoplasia. Cancer Research 56:5380-5383 (1996).
9. M. Schena, D. Shalon, R.W. Davis, and P.O. Brown. "Quantitative monitoring of gene expression patterns with a complementary DNA microarray." Science 270:467-470 (1995).
10. M.R. Emmert-Buck, C.D. Vocke, R.O. Pozzati, P.H. Duray, S.B. Jennings, C.D. Florence, Z. Zhuang, D.G. Bostwick, L.A. Liotta, and W.M. Linehan. "Allelic loss on chromosome 8p12-21 in microdissected prostatic intraepithelial neoplasia." Cancer Research 55:2959-2962 (1995).
11. Z. Zhuang, M.J. Merino, R. Chuaqui, L.A. Liotta, and M.R. Emmert-Buck. "Identical allelic loss on chromosome 11q13 in microdissected in situ and invasive human breast cancer." Cancer Research 55:467-471 (1995).
12. D. Shibata. "Extraction of DNA from paraffin-embedded tissue for analysis by polymerase chain reaction: new tricks from an old friend." Human Path. 25:561-563 (1994).
13. L.X. Pan, D.C. Diss, and P.G Isaacson. "The polymerase chain reaction in histopathology." Histopath. 26:201-217 (1995).
14. M. Loda. "Polymerase chain reaction-based methods for the detection of mutations in oncogenes and tumor suppressor genes." Human Path. 26:564-570 (1994).
15. L. Whetsell, G. Maw, N. Nandon, D.P. Ringer, and F.V. Schaefer. "Polymerase chain reaction mmicroanalysis of tumors from stained histological slides." Oncogene 7:2355-2360 (1992).