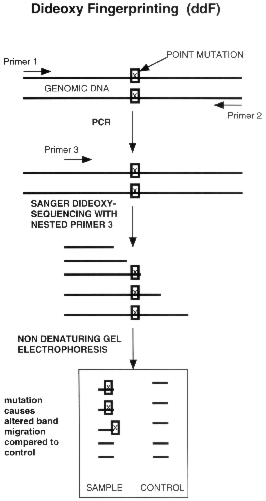
Figure 1. Schematic |
Direct genomic sequencing, the gold standard for mutation analysis, is labor-intensive, expensive, and time-consuming. Several shortcut mutation-screening methods have therefore been developed. These include heteroduplex analysis, chemical mismatch cleavage, and denaturing gradient gel electrophoresis (1). Although faster and cheaper than direct genomic sequencing, these methods are technically difficult, labor-intensive, prone to false negatives or false positives, insensitive, and may require relatively large amounts of input template DNA.
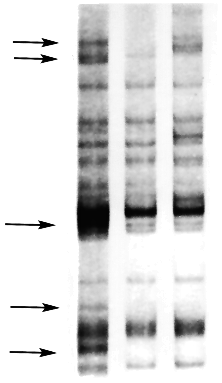
The most widely used system for detecting point mutations has been single-strand conformation polymorphism analysis (SSCP) (2). SSCP starts with PCR amplification of target DNA. The amplified and partially denatured strands are then separated on a nondenaturing polyacrylamide gel. A mutation in the DNA strand generally causes a change in the three dimensional conformation, and the altered DNA migrates to a different point on the gel compared with the wild-type control. SSCP is simple to perform; unfortunately, however, it can miss many mutations (4), such as single-base substitutions of cytosine (C) to thymidine (T), that do not alter the 3-D shape enough to significantly change the migrational properties of the strand. In addition, SSCP will provide no information about the approximate location of the mutation within the screened segment.