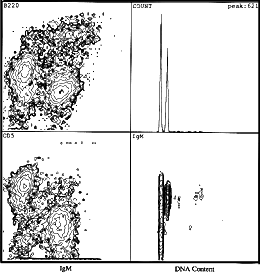
 Figure 1.
Figure 1.by Maryalice Stetler-Stevenson, M.D., Ph.D., NCI, and Gerald E. Marti, M.D., Ph.D., FDA
There are two camps when it comes to flow cytometry: the believers, who appreciate the technique as a fast lane to tomorrow's research questions, and the uninitiated, who haven't heard all the things that are possible through contemporary applications of the technique. After getting its start with the crude cell counters of the 1930s and '40s, contemporary flow cytometry is no longer just for counting. New-age flow cytometry-which includes but is not limited to fluorescence-activated cell sorting (FACS)- couples a highly sensitive, automated fluorescence-detection device with sophisticated computerized analysis of data gleaned at lightning speed for measurements of numerous interesting properties of large populations of cells. Cell size, viability-including the presence of apoptotic cells, cell- cycle dynamics, kinetics, and the presence of multiple intracellular and surface proteins can be determined for each cell in a sample.
The high sensitivity of this technique, coupled with the ability to rapidly analyze multiple parameters in samples containing 5,000 to a million cells, allows the detection and definition of unique subpopulations within a sample. The ability to detect subpopulations with an abnormal pattern of protein expression is useful in diagnosing and subclassifying leukemias and lymphomas. And because flow cytometry can routinely pick out one neoplastic cell per 1,000 normal cells-and specialized techniques can improve this sensitivity to find one abnormal cell in a million normal cells-the technique is useful in hematology and hematopathology for detecting minimal residual disease.
By using appropriate standards and controls, flow cytometry can be exquisitely quantitative, allowing researchers to determine the exact number of molecules of fluorescent antibodies-and thus the molecules of antigen-bound to a cell or within a cell. This makes flow cytometry broadly useful for precise measurement of the expression of oncogenes, activation markers, adhesion receptors, and other proteins and, depending on the method of staining, allows localization of these proteins to the cell surface or cell interior.
Flow cytometry is becoming important for exploring some of the cellular activities that are under intense research scrutiny these days. Cell-cycle data can be obtained by DNA-content analysis or by detection of bromodeoxyuridine (BrdU) incorporation or cell-cycle specific proteins. Flow cytometry is, arguably, the most sensitive and easiest method for detecting apoptosis. The TUNEL method-in which the terminal deoxynucleotidyl transferase (TdT) end- labels double-stranded DNA breaks, which are generated during apoptosis-can be applied to large populations of cells by the use of flow cytometry. Another method of flow-cytometric apoptosis detection-based on light-scatter characteristics that change when the nucleus condenses during apoptosis-requires no manipulations other than preparation of a cell suspension. Reduced DNA content, or hypoploidy, as indicated by propidium iodide (PI) staining, is another easy procedure for detecting apoptosis. Even oxidative state and the flux of ions, such as calcium, into cells can be measured by flow cytometry. The power of flow cytometry in cell biology lies not only in its sensitivity, but also in its ability to measure multiple characteristics simultaneously on each cell in a population of 100,000 to a million cells. Therefore, for example, the nonapoptotic vs. apoptotic cells in a heterogeneous sample can be rapidly compared for levels of BCL-2 and p53 protein expression, presence of a lineage-specific surface markers, such as T-cell specific antigen, and cell- cycle phase (G1/G0, S, or G2+M).
Figure 1.
Flow cytometric analysis of a murine marginal zone lymphoma.
Left panels show that the majority of the cells are positive for fluorescein
isothiocyanate (FITC)-staining of IgM, positive for phycoerythrin staining of
CD5, and positive for dim tricolor B220, which, in this case, is indicative of
marginal zone lymphoma. The right upper panel shows DNA content (x-axis) verses
cell number (y-axis). The right lower panel shows a two-parametric contour plot
of FITC IgM versus DNA content. Two G1/G0 populations can be seen; one is
diploid and the other is aneuploid. Both are IgM positive.
In flow-cytometric analysis, cells in a single cell suspension are stained with multiple fluorescent markers and transported rapidly-routinely 18,000-30,000 cells/min -to intersect a finely focused monochromatic beam of light of an appropriate frequency. A stream of fluid containing the sample cells is ejected at steady pressure and rate into a flowing, high- pressure "sheath" fluid. The convergence of the sample stream with the sheath fluid allows for the precise intersection of the sample stream that contains the single cell suspension with the laser beam. This is referred to as hydrodynamic focusing. The fluorochromes attached to the cells absorb light and emit energy at a longer wavelength that is specific for the fluorochrome. For example, fluorescein isothiocyanate (FITC) emits light at a different wave length than phycoerythrin (PE) or peridin-chlorophyll-a- protein (PerCP), allowing all three indicators to be detected simultaneously in a cell. In addition, light is scattered in proportion to the size of the cell in the forward direction, much like a shadow. Light is also scattered or reflected to the side by the intracellular granules; thus, cells with complex cytoplasm, such as the granular neutrophils, have a much higher side scatter than do less complex cells, such as lymphocytes.
Light detectors collecting the forward and side scatter and fluorescence emissions thus rapidly gather information for each cell on its size, cytoplasmic complexity, and fluorescent markers. Three different fluorochromes emitting light at three different frequencies are routinely used in clinical laboratories, whereas research facilities may use five or more fluorochromes. Fluorochromes may be bound to antibodies for the detection of specific proteins or of BrdU incorporation in studies of cell- cycle kinetics. Alternatively, the fluorochromes may be used for direct staining of cellular elements. An example of this would be PI intercalation into DNA.
Flow cytometry measures all parameters for all cells passing through the focused beam, and then the cells can be classified into categories based on any combination of the detected parameters, including presence of cell- surface lineage-specific markers or activation antigens, oncogene expression, or DNA content. Complex computer programs then quantify the numbers of cells within each defined category and record the fluorescent intensity, indicating levels of protein expression or quantities of DNA, for example. The computer then displays the data in two- or three- dimensional plots of parameters and calculates the percentage of cells falling into any category specified by the investigator.
Mention of a specific product in the following description does not constitute an endorsement.
Protocols differ depending on the information a researcher is seeking- cell-cycle analysis or cell-surface antigen, for example-and the type of specimen-cell lines in media, whole blood, or bone marrow, for example. The two most widely used protocols are whole-blood lysis (WBL), for surface immunophenotyping of blood or bone marrow, and PI staining of isolated cells, for analyzing the cell-cycle (to detect S phase cells) and for detecting aneuploidy.
In WBL, a 10- to 100-mL sample of whole blood or bone marrow is stained with two to five conjugated monoclonal reagents. After the staining is complete, the red cells are lysed using either dilute HCl, hypotonic solution, detergents, ammonium chloride, or proprietary preparations. The lysed cells are washed and fixed for analysis. Compared with mononuclear cell isolation followed by antibody staining, WBL is more convenient, quicker, and less likely to cause selective cell loss, which could bias the data. Fixation stabilizes cell membranes, crosslinks the antibody to its antigen, and reduces the risk that researchers will be contaminated with infectious material. Fixed cells are also stable for longer periods than are fresh cells.
To stain intracellular antigens, the cells are permeabilized and stained with conjugated monoclonal antibodies. Red cells are lysed. If the permeabilization method does not fix the cells, they must be fixed after lysis of the red cells. For simple cell-cycle analysis, cells are permeabilized and stained with a DNA dye such as PI. Combined antigen- expression and DNA-content analysis usually consists of staining with an FITC-labeled antibody, followed by gentle fixation, permeabilization, and PI staining. Both FITC- and PE-labeled reagents can be used together with 7- amino-actinomycin D (7AAD) instead of PI, which emits light in the same wavelength region as PE. Describing the actual acquisition and computer analysis of the data on the flow cytometer is beyond the scope of this article. In our opinion, acquisition and analysis of flow-cytometry data is best done by-or under the supervision of-someone experienced in the field. Labs with substantial flow-cytometry experience and facilities are listed in the "contacts" section of this article.
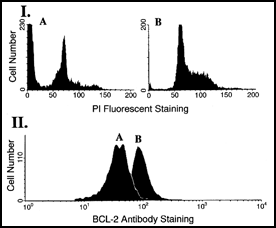 Figure 2.
Figure 2.
I. DNA content analysis to detect apoptosis in cells after cold shock.
Cell line A is prone to apoptosis in response to cold shock.
Sub-diploid apoptotic peak is shown. Cell line B is resistant to cold
shock-induced apoptosis and shows no sub-diploid apoptotic peak.
II. BCL-2 expression in cell lines.
Apoptosis-resistant cell line B has higher level of BCL-2 protein expression than
apoptosis-prone cell line A.
Attention to several technical details helps to prevent problems and to identify the source of problems that do occur. We wash all cells that are derived from living animals, including humans, to remove adherent proteins that may bind labeled antibodies or dyes in a specific or nonspecific manner. Using too high a concentration of antibody promotes nonspecific binding. The appropriate concentrations for antibodies are often specified by manufacturer or can be empirically determined by staining control cells with serial dilutions. When working with antibodies, we use isotypic controls against nonmammalian proteins to detect nonspecific binding of antibodies. Negative and positive controls for all biological processes that are being characterized in a flow-cytometry study should be run to ensure sensitivity and specificity. Protocols of preparations for flow- cytometric analysis of cell-surface, intracellular, and intranuclear antigens and DNA content are given below.
Add 20 mL or less of peripheral blood, bone marrow, or cell suspension to a 50-mL centrifuge tube. With peripheral blood or bone marrow, mark the level.
Add room temperature phosphate-buffered saline (PBS) to bring the volume to 45 mL. Invert to mix and centrifuge at 1200 rpm in swinging- bucket rotor centrifuge at room temperature for 10 min.
Aspirate the supernatant and repeat step 2. above two times.
Restore to original volume with PBS. Dilute to 2 x 10^6 cells/mL.
Add appropriate amount of antibodies to labeled tubes (usually 5-20 microliters). Three antibodies, each complexed with a different color fluorochrome (e.g. FITC, PE, and PerCP) can be placed in the same tube for FacScan analysis. For five-color analysis or UV excitation, more complex flow cytometers are required. Add 150 microliters PBA (PBS with 0.1% bovine serum albumin and 0.1% NaN3) or PBS with 10% fetal calf serum (FCS) to each tube.
Add 100 microliters of washed whole blood or bone marrow to each of the prepared reagent tubes and incubate in the dark for 30 min at room temperature. Because light bleaches fluorochromes, tubes should be kept covered with aluminum foil.
Wash cells by adding 4 mL PBA and centrifuging at 1200 rpm for 8 min in a swinging-bucket rotor centrifuge at 4-6 degrees Celsius. Aspirate the supernatant. Approximately 100 microliters PBA and cells will remain.
Lyse the specimens using a proprietary lysing kit (e.g., Immunolyse, Q-prep, and Facslyse). We find that manually lysing is quickest and provides an excellent specimen in the hands of an experienced technician. The manual lysing methods vary from product to product but usually entail:
Timing, in manual lysing, is critical. We recommend using an electronic timer and restricting the number of tubes to be lysed to a number that can be handled promptly. Experienced technicians in our laboratory do not lyse more than 20 tubes in one batch. Machines that perform the entire lysing procedure (e.g., the Coulter Q-prep) are available but can lyse just one tube at a time. The main benefit of the Q-prep is that it requires no experience to produce an excellent specimen. Its main drawback is that it is not as fast as manual lysing in the hands of an experienced technician.
After washing the cells with PBA, resuspend each specimen while vortexing lightly in 500 microliters of 1% paraformaldehyde in the isotonic solution that the cells will be analyzed in. Cover and keep at 4-6 degrees Celsius in the dark for at least 1 h but, preferably, overnight.
Place 5-20 mL of PE-conjugated monoclonal antibody for the detection of the antigen of interest (e.g., CD3 for T cells) into labeled, flow- cytometer-compatible tubes. Add 150 mL PBA and 100 mL washed cells. Incubate for 30 min at room temperature in the dark.
Add 2 mL 1x Ortho PermaFix* to each tube, vortex for a second or two, and incubate for 40 min at room temperature in the dark.
Centrifuge for 8 min at 1400-1600 rpm in a swinging-bucket rotor centrifuge at 4-6 degrees Celsius. Aspirate off the supernatant and vortex the pellet thoroughly.
If specimen is whole blood or bone marrow, lyse the red blood cells by adding 2 mL PBS, vortexing thoroughly, and incubating at room temperature for 10 min in the dark. Vortex again and centrifuge for 8 min at 1400-1600 rpm in a swinging-bucket rotor centrifuge at 4-6 degrees Celsius. Decant supernatant and vortex gently.
Add 20 mL appropriately diluted antibody [e.g. TdT or myeloperoxidase (MPO)] to tubes and vortex lightly. Incubate in an ice bath for 1 h in the dark.
Wash cells by adding 2 mL PBA, centrifuging at 1400-1600 rpm in a swinging-bucket rotor centrifuge at 4-6 degrees Celsius, and decanting supernatant.
Resuspend pellet in 0.5 mL 1% paraformaldehyde in isotonic saline, pH 7.4. Analyze samples on flow cytometer within 24 hours. Other permeabilization reagents can be used, but method must be optimized for each reagent.
For whole blood and bone marrow, begin by separating mononuclear cells by density gradient (e.g., by using Ficoll Hypaque). Wash this cell suspension or others, such as cells from a cultured cell line or cells teased from lymph node, with PBS prior to staining.
Place 100 mL PBS in labeled, flow-cytometer-compatible tubes and add an appropriate amount of FITC-conjugated antibody for detection of the surface antigen of choice. Add one million to two million cells in 100 mL. Incubate in the dark at 4-6 degrees Celsius for 30 min.
Wash by adding 4 mL cold PBS (4-6 degrees Celsius), centrifuging at 1200 rpm in swinging-bucket rotor centrifuge at 4-6 degrees Celsius for 10 min, decanting supernatant, and resuspending pellet by agitating tube or light vortexing. Repeat wash.
Add to resuspended cells 1 mL 70% cold (4-6 degrees Celsius) ethanol per 1 x 106 cells while vortexing gently. Incubate in the dark overnight at 4-6 degrees Celsius to fix cells.
Wash by adding 4 mL cold PBS (4-6 degrees Celsius), centrifuging at 1600 rpm in swinging-bucket rotor centrifuge at 4-6 degrees Celsius for 5 min, decanting supernatant, and resuspending pellet by agitating tube or by light vortexing.
Add 500 mL PI/RNase solution (50 mg/mL PI, 200 units RNase/mL in PBS). Incubate 45 min to 1 h at room temperature. Analyze samples on flow cytometer within 4 h.
H.M. Shapiro. "How a Flow Cytometer Works." In: Practical Flow Cytometry 2nd ed. H.M. Shapiro, ed. New York: Alan R. Liss. (1994).
R.W. Vogt, Jr., G.E. Marti, and A. Schwartz. "Quantitation calibration of fluorescence intensity for clinical and research applications of immunophenotyping by flow cytometry." In: Reviews of Biotechnology and Bioengineering. vol. 1, H.W. Tyler, ed. Norwood, N.J.: Ablex Publishing Corp. (1995).
M.A. Moltz, J. Gong, F. Taraganos, and Z. Darzynkiewicz. "Flow cytometric detection of apoptosis: comparison of the assays of in situ DNA degradation and chromatin changes." Cytometry 15, 237-44 (1994).
J. Gong, F. Traganos, and Z. Darzynkiewicz. "A selective procedure for DNA extraction from apoptotic cells applicable for gel electrophoresis and flow cytometry." Analy. Biochem. 218, 314-19 (1994).
L.L. Wheeless, J.S. Coon, C. Cox, A.D. Deitch, R.W. deVere White, Y. Fradet, et al. "Precision of DNA flow cytometry in inter-institutional analysis." Cytometry 12, 405-12 (1991).
R.P. Wersto, R.L. Liblit, and L.G. Koss. "Flow cytometric DNA analysis of human solid tumors: a review of the interpretation of DNA histograms." Human Pathol. 22, 1085-98 (1991).
M. Stetler-Stevenson, L.J. Medeiros, and E.S. Jaffe. "Immunophenotypic methods and findings in the diagnosis of lymphoproliferative diseases." In: Surgical Pathology of the Lymph Nodes and Related Organs. 2nd ed. Philadelphia: W.B. Saunders Co., pp. 22-57 (1995).
R.P Wersto and M. Stetler-Stevenson. "Debris compensation of DNA histograms and its effect on S-phase analysis." Cytometry 20, 43-52 (1995).