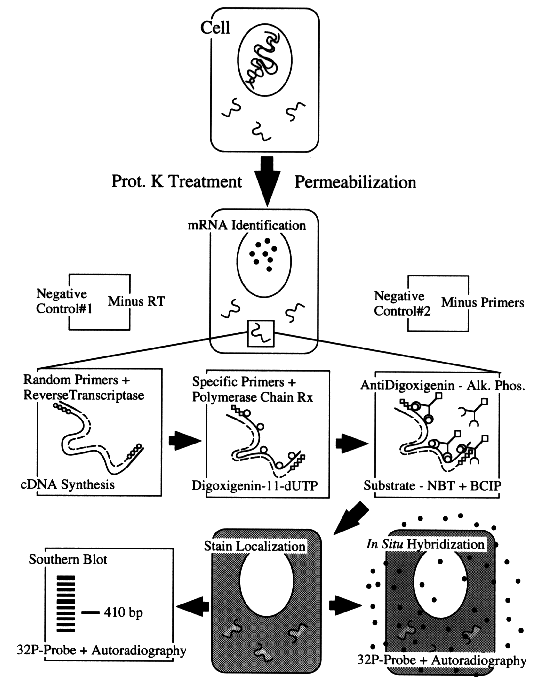
Figure 1. Schematic drawing of the steps involved in the amplification of mRNA in tissue sections. The basic controls have been also included.
by Alfredo Martínez, Ph.D., NCI, and Frank Cuttitta, Ph.D., NCI
The possibility of performing DNA or mRNA amplification in tissue sections has been proposed since the beginning of the polymerase chain reaction (PCR) era. Unfortunately, however, the technique has proved to be more elusive than a quick glance of various protocols (1,2) suggests. Nevertheless, the potential reward promised to those able to master the technique is high: unlimited sensitivity in the detection of specific nucleic acids expressed in subpopulations of cells with as little as a single "available" molecule inducing a detectable signal. This promise has attracted a great number of investigators who work with molecules that are expressed at low levels (e.g., growth factors, receptors, and developmental signals), with new or rare genes (e.g., viral infections, point mutations, transpositions, and deletions), or who are trying to follow vectors after gene therapy.
The advent of thermocyclers designed specifically for tissue sections solved many of the technical problems associated with detecting these low-level signals in preserved tissue, and our laboratory recently developed a direct protocol that uses one such instrument to detect DNA and mRNA in archival material (3).
Figure 1. Schematic drawing of the
steps involved in the amplification of mRNA in tissue sections.
The basic controls have been also included.
The Method and How It Works
In the past, one of the most serious problems with in situ PCR was the lack of reproducible results due to 1) difficulties in performing synchronized "hot start" applications, 2) limitations in the number of slides that can be processed at the same time, and 3) heterogeneous heating of the slides (they were usually placed on top of a regular thermocycler block, with holes for the tubes, and even if a small aluminum foil boat is used, there can be large temperature variations in different areas of a tissue section). We overcame the first obstacle by using a monoclonal antibody that blocks Taq polymerase until the temperature reaches 70°C. At that temperature, the denatured antibody liberates an active enzyme to the PCR solution (4). The other two difficulties were resolved by performing the reaction in a thermocycler specifically designed to accommodate microscope slides (Hybaid's OmniSlide System, National Labnet Company, Woodbridge, N.J.).
In addition to these problems, fixation of tissue is critical for obtaining good results in in-situ PCR. In samples treated with alcohol- or acetone-based fixatives, the PCR products ended up in the supernatant. Conversely, cross-linking fixatives, such as paraformaldehyde or formalin, retain the labeled products in the tissue, possibly by entrapping them in the lattice they created among proteins (5).
Conceptually, the protocol is simple (see figure 1). If we want to detect DNA, after dewaxing and rehydrating the sections, three steps must be performed: 1) protein digestion to facilitate reagent penetration, 2) PCR reaction with simultaneous labeling of the PCR products, and 3) visualization of the labeled products by immunocytochemical methods. The detection of mRNA incorporates a reverse-transcription step, to generate cDNA, before amplification.
In situ amplification combines all the advantages of histological and PCR technologies but, unfortunately, it also compounds the possible artifacts of both. For this reason, successful interpretation of results requires attention to appropriate controls (see figure 2). These are some of the controls we use: 1) positive control - a section of a tissue or cell line known to have a high expression of the target nucleic acid as determined by other techniques (such as Northern blot, regular PCR, or in situ hybridization); 2) negative control - substitution of primers by water in the PCR mixture to reveal non-specific endogenous priming (from necrosis, apoptosis, or repair processes, for example); 3) negative control for mRNA - use RNase pretreatment or omit reverse transcriptase; 4) co-localization of the signals for the mRNA - via in situ RT-PCR - and, for its translated protein, via immunocytochemistry; 5) extraction of the DNA from the tissue section after amplification and analysis by electrophoresis and Southern blot; and 6) in-situ hybridization with a labeled nested probe after amplification. The last procedure is routinely used in the indirect method of in situ PCR.
Protocol
In situ amplification can be done on cytospin preparations or on sections from paraffin-embedded and frozen tissues. Here we present a protocol for the detection of mRNA in paraffin sections that is important because of its application to archival material. To detect DNA, step 4 would be omitted. Mention of specific products does not constitute an endorsement.
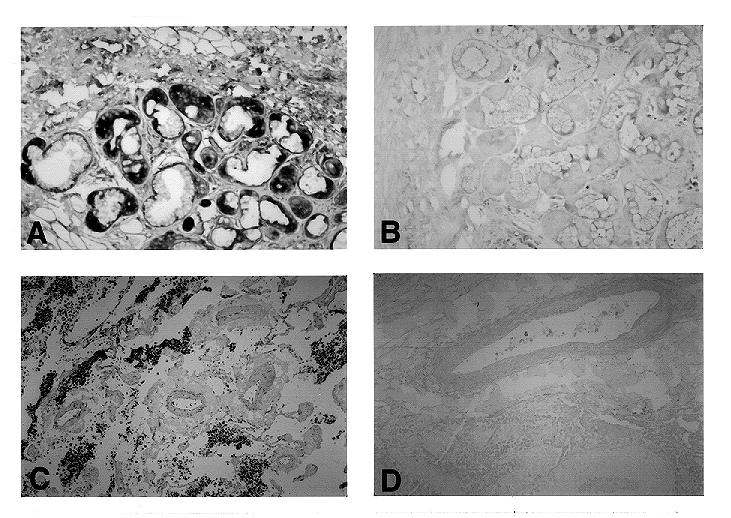
Figure 2.Detection of mRNA in archival
material by in situ RT-PCR. a) Localization of adrenomedullin
mRNA in human bronchial glands. This regulatory peptide has been
recently described in normal lung and in pulmonary tumors (6).
b) Serial section of the same gland used as a negative control,
primers were substituted by water in the PCR mixture. c) Large
cell pulmonary tumor expressing transferrin mRNA (7). d) Negative
control obtained by omission of the reverse transcription.
In Situ RT-PCR
1. Take the usual precautions for working with RNA, even during cutting and handling of sections: wear gloves, bake the glassware, and use diethyl pyrocarbonate (DEPC)-treated water.
2. Deparaffinize the sections by immersion in xylene (20 min.) then rehydrate in decreasing concentrations of ethanol in DEPC-treated water. Always use new solutions for in situ RT-PCR. You can subsequently reuse these solutions for regular histological procedures.
3. Permeabilize the tissue by incubation with proteinase K. A concentration of 10 µg proteinase K/mL at 37°C for 15 min. is appropriate for most archival material, but varying the concentration or exposure time is recommended to optimize results for each particular application.
4. For reverse transcription, prime the reverse transcriptase. For this step, you may use either specific primers designed to target the proper message or an oligo (dT) that binds to the polyA tail of the mRNAs. We use the SuperScript Preamplification System (Gibco BRL, Gaithersburg, Md.): first, a drop (60 µL) containing the primers is placed on top of the section and covered with a coverslip of parafilm, then the sections are incubated for 10 min. at 70°C in the thermocycler. After removing the coverslips, another solution containing the reverse transcriptase (100 U/section) is added and covered with a new piece of parafilm. The slides are then maintained at room temperature for 10 min., at 45°C for 45 minutes, and at 70°C for 10 minutes.
5. For PCR, optimize all the parameters (such as pH, MgCl2 concentration,
and annealing temperature) for each specific set of primers used
in the PCR reaction by regular PCR before attempting in situ amplification.
a) Mix in a sterile microcentrifuge tube 0.5 µL of Taq polymerase
(Perkin-Elmer Cetus, Norwalk, Conn.) and 0.5 µL of TaqStart
antibody (Clontech, Palo Alto, Calif.) per slide and incubate
5 min. at room temperature to block the enzyme. b) Add the rest
of the components of the PCR mixture to obtain the following composition:
2.5 mmol MgCl2/L, 200 mmol dNTPs/L, 100 µmol digoxigenin-11-dUTP/L
(Boerhinger Mannheim, Indianapolis), 1 ng primers/µL, 50
mmol KCl/L, 10 mmol Tris-HCl/L. c) Apply 80 µL of solution
to each slide, then cover the section with a glass coverslip and
seal with rubber cement to prevent evaporation. Place the slides
in the thermocycler. d) Optimize the number of cycles and the
annealing temperature for each tissue and target nucleic acid.
A standard run could be like this: begin with 2 min. at 72°C;
15 cycles: 94°C for 15 s., 55°C for 15 s., 72°C
for 60 s.; finish with 5 min. at 72°C.
e) Remove coverslips and wash the sections twice - for 20 min.
each time - in 0.1X SSC at 45°C.
6. For detecting digoxigennin-tagged DNA, we use the Digoxigenin Detection kit (Boehringer Mannheim). To produce a dark blue precipitate, it requires a 2-h. incubation with an antidigoxigenin antibody bound to alkaline phosphatase at a dilution 1:500, thorough washes, and incubation with the proper substrates (nitroblue tetrazolium and a complex phosphate).
7. Check slides under the microscope until the proper color intensity is reached. Stop the reaction before the background in the negative controls begins to increase. Mount the slides in a water-soluble mounting medium (such as Crystal Mount, Biomeda, Foster City, Calif.) because the blue precipitate is soluble in alcohols.
8. Compare the test slides with the controls.
Troubleshooting Tips
1. How to choose a thermocycler? Several companies offer slide thermocyclers for in situ PCR with different designs. Before buying one of them, look for the following characteristics: a) number of slides that can be processed at the same time - remember, the name of the game is controls - and the more sections you can accommodate per run, the better; b) ease of operation; try to avoid complex manipulation; c) price, which is unquestionably a critical point and a variety of instruments are available (e.g., Perkin Elmer, Coy, MJ Research, and Hybaid), costing between $3,000 and $10,000; and d) maintenance service support following sale (e.g., is there a regional distributor in the area?).
2. DNase or not DNase? In some protocols for in situ RT-PCR, the authors recommend a digestion with RNase-free DNase before the reverse transcription step. This treatment is intended to remove nuclear and mitochondrial DNA to avoid genomic amplification during PCR. We repeatedly observed, using a variety of DNases, that this digestion step resulted in nonspecific nuclear staining (3). This problem seems to be due to the behavior of the DNase enzyme, which cuts the DNA into oligonucleotides but does not reduce it to mononucleotides. The remaining oligonucleotides are used as primers by the Taq polymerase, leading to nonspecific staining. For this reason, we strongly recommend omission of this step. A careful choice of primers and a reduced number of cycles (15 to 20) helps to avoid nonspecific nuclear staining.
3. Designing primers. When choosing primers, consider the following: a) A good size range for the PCR product is 100 to 500 base pairs (bp). If the product is too small, it could leak out of the fixative-induced lattice and be washed out of the tissue. On the other hand, excessively long products could be hard to amplify in tissue sections, and, especially in archival material, the probability of finding nicks in the nucleic acid template that prevent amplification increases with size. b) If primers bridge an intron, it helps to eliminate the possibility of genomic amplification. c) Take precautions to avoid palindromic sequences and hairpin formations in the primers. These formations can block amplification.
4. Double labeling. It is possible to combine in situ amplification with immunocytochemistry or in situ hybridization. For immunocytochemistry, we recommend performing immunological detection first because thermal cycling could destroy antigens in tissue. For mRNA localization, remember to use RNase-free reagents during the whole process by using DEPC-treated water in all solutions and adding RNase inhibitors to antisera.
Contacts:
Alfredo Martínez, NCI
Phone: 402-3128
e-mail: martineza@bprb.nci.nih.gov
Masahito Ebina, NCI
Phone: 402-3128
e-mail: ebinam@bprb.nci.nih.gov
References
1. M. Corcoran, M. Levin, S. Jacobson, and L. Liotta. "From hot starts and false starts to smart starts: in situ PCR." The NIH Catalyst 2, 12 - 14 (1994).
2. G.J. Nuovo. "PCR in situ hybridization: protocols and applications." New York: Raven Press (1992).
3. A. Martínez, M.J. Miller, K. Quinn, E.J. Unsworth, M. Ebina, and F. Cuttitta. "Non-radioactive localization of nucleic acids by direct in situ PCR and in situ RT-PCR in paraffin-embedded sections." J. Histochem. Cytochem. (in press).
4. D.E. Kellogg, I. Rybalkin, S. Chen, N. Mukhamedova, T. Vlasik, P.D. Siebert, et. al. "TaqStart antibody; "hot start" PCR facilitated by a neutralizing monoclonal antibody directed against Taq DNA polymerase." BioTechniques 6, 1134 - 37 (1994).
5. J.J. O'Leary, G. Browne, R.J. Landers, M. Crowley, I. Bailey Healy, J.T. Street, et. al. "The importance of fixation procedures on DNA template and its suitability for solution-phase polymerase chain reaction and PCR in situ hybridization." Histochem. J. 26, 337 - 46 (1994).
6. A. Martínez, M.J. Miller, E.J. Unsworth, J.M. Siegfried, and F. Cuttitta. "Expression of adrenomedullin in normal human lung and in pulmonary tumors." Endocrinology (in press).
7. M.J. Miller, K. Quinn, M.D. Vos, A. Treston, E.J. Unsworth, J.L. Mulshine, et. al. "Expression of transferrin mRNA in human tumor cell lines." Proc. AACR 35, 552 (1994).