HIV-1 INDUCES A NEW, G2-PHASE FORM OF CELL DEATH IN T-CELLS
by David Cohen , M.D.,
of the Laboratory of Immunoregulation, NIAID. Cohen presented
this lecture on March 29, 1995, as part of the Immunology Interest
Group's regular seminar series.
Abstract
Human immunodeficiency virus - type 1 (HIV-1) infection in humans
leads to depletion of CD4+ T-cells and the development of AIDS.
Despite intense investigation over the past decade, the mechanisms
underlying CD4+ cell death in HIV disease are poorly understood.
Genetic-mapping studies of HIV have provided some insight into
this process by showing that the most important determinants of
the virus' ability to kill T-cells lie within the viral envelope
glycoproteins (gp120 and gp41). In the past several years, it
became increasingly clear that programmed cell death, or signaling-dependent
cell death, plays a major role in many forms of physiologic and
pathologic cell death. We and others asked whether the form of
programmed cell killing accounts for HIV's cytopathicity. Our
experiments suggest that the virus invokes a distinct form of
programmed cell death that kills T-cells in the G2 phase of the
cell cycle.
A new, pathologic type
of programmed cell death, dubbed "phouskomatosis"-coined
from phouskoma,
the Greek word for bloated or inflated, is triggered by
HIV infection and kills T-cells in the G2 phase of the cell cycle.
HIV acts at the G2 checkpoint via regulatory proteins such as
mos and abl. In contrast, classic apoptotic cell death occurs
when T-cells are in the G1 phase and operates through regulatory
proteins such as p53, Myc, and Ras.
Questions
Q: What was the starting point for
this work?
A: We initially investigated whether any
of the proteins encoded by HIV-1 were capable of initiating intracellular
signals that directly program CD4+ cells to die. These studies
led us to conclude that processed HIV envelope glycoproteins (gp120
and gp41) expressed on the surface of one T-cell can interact
with the CD4 receptor of another T-cell, triggering signaling
mediated by protein tyrosine kinases (PTKs) and cell death. Other
HIV proteins, including Tat, Rev, Nef, and the matrix polypeptides,
are not capable of directly initiating CD4+ cell death. Using
the PTK inhibitor herbimycin A, we also showed that interfering
with protein tyrosine phosphorylation during HIV infection dramatically
reduces viral cytopathicity in vitro.
We followed up this initial observation by attempting to identify
the viral and cellular substrates that undergo tyrosine phosphorylation
during the course of HIV infection. Our observations with antiphosphotyrosine
antibodies suggest that a 34-kilodalton cellular substrate becomes
profoundly tyrosine-hyperphosphorylated and that this event has
the same kinetics as HIV-induced cell death. We also found that
an HIV matrix protein (p17 gag) may also become tyrosine-phosphorylated
during the course of HIV infection.
To define these phosphorylated substrates more completely, we
performed phosphoamino acid analysis, which showed that the cellular
substrate had the unusual property of being phosphorylated at
threonine and serine residues as well as at a tyrosine residue.
This suggested to us that the pp34 substrate might be a cyclin-dependent
kinase (cdk), which was verified when we established that the
pp34 substrate is cdk1, the cdk regulator of G2/M transition.
Q: Which findings have been most
surprising to you or to other scientists?
A: The identification of large quantities
of tyrosine-phosphorylated cdk1 in cells that were dying during
HIV infection strongly suggested that these cells were trapped
in the G2 phase of the cell cycle, where tyrosine-phosphorylated
cdk1 accumulates. This observation was surprising because all
previously described forms of apoptotic cell death in normal T-cells
had been shown to occur when the cells were in G1 or early S phase.
We confirmed that observation through additional biochemical experiments,
including analysis of the mitotic cyclin, cyclin B. These studies
also showed that clinical isolates of HIV-1 that have the greatest
cell-killing capacity also most strongly direct the G2 form of
cell death. This correlation makes this killing pathway a focal
point for understanding depletion of CD4+ T-cells in HIV-1 infection.
Q: What were the greatest stumbling
blocks, and what new observations, techniques, reagents or insights
helped you to get past them?
A: Because HIV-1 infection or processed envelope
glycoproteins (gp120 and gp41) might also be capable of triggering
G1/S forms of apoptosis or might initiate cell death by additional
mechanisms such as syncytium formation, it was important to distinguish
the pathway that we had identified from these other processes.
To overcome this difficulty and to define the G2-cell-death pathway,
we eluted dying cells and fractionated a purified population of
"balloon" cells. These dying "balloon" cells
have a single, open nucleus, are free of syncytia, and show no
signs of classical apoptosis, which is characterized by pronounced
nuclear condensation.
Transmission electron microscopy studies of purified balloon cells
tagged with immunogold demonstrated that the balloon cells have
active centrosomes, containing both cyclin B and cdk1 proteins,
which again confirmed that these cells are trapped in G2 - the
part of the cell cycle in which centrosomes become activated.
Q: Do you see any potential areas
where this research might provide insights for clinical scientists,
and how are you following up on your discoveries?
A: These findings have general significance for
understanding pathological forms of programmed cell death and
may also provide therapeutic targets in the cell for inhibiting
HIV-1 directed T-cell killing. Our studies suggest that HIV-1
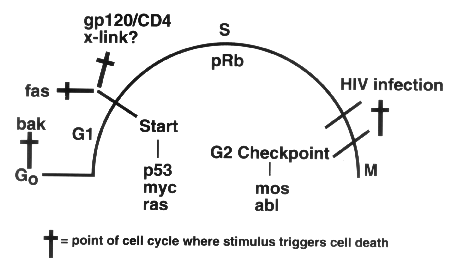
infection initiates a pathological form of cell signaling leading
to prominent death of cells at G2 (see figure, page 8). In contrast,
physiologic forms of T-cell death, and quite possibly other pathways
of cell killing triggered by HIV-1 (see figure, page 8), occur
at G1, where induction and subsequent cross-linking of the Fas
apoptotic receptor is an important mediator of T-cell death. We
expect that genes involved in S and G2 cell-cycle progression
are likely also to be responsible for initiating and executing
the HIV/G2 cell death programs in the balloon cells (see figure,
page 8). Such genes include pRb, c-mos, and c-abl rather
than other cell-cycle regulatory genes, such as p53, myc,
and ras, which appear to mediate apoptosis in G1. Because
the G2 program is rarely employed during normal T-cell regulation,
therapeutic intervention in the G2 pathway might interrupt CD4+
T-cell depletion during HIV infection without interfering with
normal T-cell function, and we are currently investigating this
interesting possibility.
Table of Contents