A hypothetical SLPI defense against HIV-1
by Sharon M. Wahl, Ph.D., Chief, Cellular Immunology Section, Laboratory of Immunology, NIDR; Tessie B. McNeely, Ph.D., Cellular Immunology Section, Laboratory of Immunology, NIDR; and Stephen P. Eisenberg, Ph.D., Synergen Inc., Boulder, Colo.
A hypothetical SLPI defense against HIV-1
Over the past decade, the absence of epidemiological evidence for adult oral transmission of HIV-1 has intrigued investigators. Initially overlooked was the possibility that the host has, in its defensive repertoire, mechanisms to stave off oral retroviral invasion. The near absence of orally transmitted HIV-1 -- like the failure of some individuals to become infected despite repeated exposure to the virus and the emerging profile of seropositive long-term survivors and nonprogressors (1,2) -- may provide us with several important opportunities to pursue and identify endogenous host mechanisms of defense against retroviruses. Within the oral cavity, a small endogenous protein may be one source of this antiviral defense, which may account, at least in part, for protection against HIV-1 shed into the oral cavity or acquired through oral exposure (3). Because AIDS is considered to be primarily a mucosally transmitted disease, this mucosa-specific inhibitor may contribute to an initial line of defense.
More than 20 years ago, a low-molecular-weight proteinase inhibitor was described in bronchial secretions (4) and named antileukoprotease (ALP) because of its ability to inhibit granulocyte proteinases (5). Similar serine proteinase inhibitors were subsequently found in other mucous secretions, including those of salivary glands (6,7). Given the widespread distribution of this antiprotease in mucosal fluids and its elastin-protecting capacity, researchers considered its primary role to be protection of the parenchyma against leukocyte proteolytic attack.
In 1986, Robert Thompson of Synergen and Kjell Ohlsson of the University of Lund, Sweden, purified this protease inhibitor from large volumes of parotid secretions, and the newly sequenced protein was dubbed secretory leukocyte protease inhibitor (SLPI) (8). Once SLPI's amino acid sequence was known, various protease inhibitors identified at other mucosal surfaces were found to be identical to the nonglycosylated polypeptide with a molecular weight of 12 kDa and an isoelectric point (pI) greater than 9 (8). The protein, shaped like a boomerang (9), consists of two homologous cysteine-rich domains of 53 and 54 amino acids, which are encoded on separate exons (10). Produced by cells of mucosal surfaces, SLPI is a potent inhibitor of human neutrophil elastase and cathepsin G, and it also inhibits other serine proteases, such as trypsin and chymotrypsin. The recombinant protein (rSLPI), produced in Escherichia coli, has the same amino acid sequence, composition, and activity as the native molecule. Researchers at Synergen have shown by site-directed mutagenesis that residue Leu-72 within the COOH-terminal domain is the active site for inhibition of leukocyte elastase and cathepsin G, as well as of chymotrypsin and trypsin (11). The NH2-terminal domain stabilizes and enhances the activity of the protease-inhibitor complex (12).
Five years after the initial purification and sequencing of SLPI, as scientists in our lab were looking for a mysterious HIV-inhibiting fraction that we had isolated from saliva, our path crossed with the path of researchers exploring SLPI's potential as a therapeutic antiprotease in lung disease. Thus, in 1991, we began our experiments in which SLPI was tested and shown to have surprising efficacy at inhibiting HIV-1 infection in vitro. At physiologic concentrations (~1 ug/mL), SLPI inhibits the appearance of reverse transcriptase (RT) activity in human monocyte/macrophage cell cultures exposed to HIV-1 (3). Although the mechanism has not been fully deciphered, the SLPI inhibition is remarkably long-lasting: a single one-hour SLPI treatment at the time of infection suppresses RT activity through several weeks of culture. In addition to inhibiting HIV in monocytes, SLPI inhibits infection of T cells and T-cell lines by laboratory and clinical HIV isolates (3, T.B. McNeely, S.P. Eisenberg, D. Dripps, and S.M. Wahl, unpublished observations).
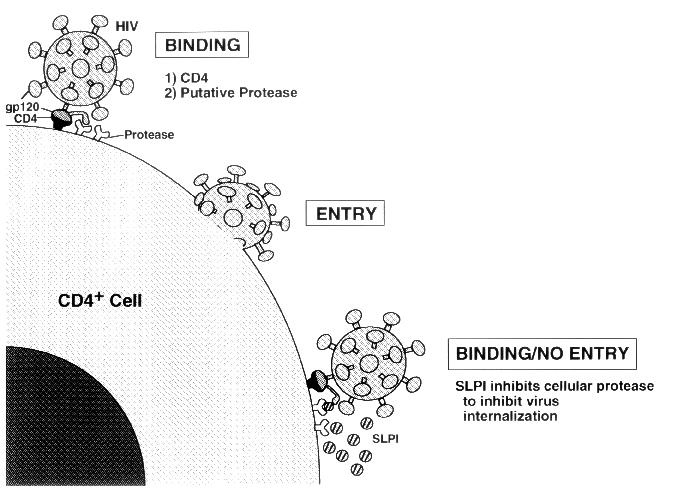
Another key observation we made in vitro was that cells pretreated with SLPI and washed prior to exposure to HIV-1 were still protected, whereas pretreatment of the virus with SLPI was not inhibitory. This observation has led us to hypothesize that SLPI inhibits HIV by acting on some target in or on the cell -- not the virus. This hypothesis was supported by our inability to demonstrate any interaction between SLPI and purified viral components, including gp120, gp160, or HIV aspartyl protease. Importantly, if SLPI's inhibition of HIV is, indeed, related to some cellular molecules and processes, these may ultimately provide a more stable target for the design of anti-HIV therapies than the elusive, rapidly mutating virally encoded proteins that form the basis for some current therapies and candidate vaccines, for example.
Armed with these in vitro observations, we are trying to piece together a hypothesis of how and when SLPI acts during HIV's infection-replication cycle to foil the virus. As is true for many viruses, HIV replication requires internalization of infectious virions and subsequent utilization of host cellular machinery for the production and assembly of new infectious particles. Antiviral agents that inhibit binding of the virion to the target cell may be removed after initial infection has been averted. On the other hand, compounds such as AZT and R031-8959 that work by disrupting the action of the viral enzymes have no activity during internalization and must be available intracellularly during proviral formation (13) or during the assembly of new infectious particles (14).
Our suspicions are thus that SLPI exerts its anti-HIV activity primarily, although perhaps not exclusively, during internalization. Like recombinant, soluble CD4, a competitive inhibitor of HIV binding, SLPI's inhibitory activity only requires it to be present while the virus attempts to dock on the target cell. Unlike CD4, however, SLPI does not bind viral-coat compounds, but it does bind specifically and with high affinity (1-10 nmol/L) to intact monocytes and to T-cell and monocyte-cell lines (T.B. McNeely, S.P. Eisenberg, D. Dripps, and S.M. Wahl, unpublished observations).
This leads us to the consideration of what cellular components SLPI interacts with to inhibit HIV penetration of the cell, and a few emerging observations from HIV studies have suggested some possibilities. Researchers now believe that HIV binding to CD4 is necessary for penetration of T cells and monocytes but suspect that CD4 binding may not be sufficient for viral entry into targeted cells. Nevertheless, attempts to identify an accessory molecule enabling HIV entry have been frustrating and in-conclusive. Among the list of candidate accessory molecules are several proteolytic enzymes. These enzymes are considered possible candidates because cleavage of HIV's V3 loop of the envelope glycoprotein gp120 is purported to be necessary for viral internalization (15) and because the loop is readily clipped by several proteases common to the white cell.
The possibility that proteolytic cleavage of HIV's coat protein is required after the virus binds to CD4 points to a potential role for SLPI: blocking this cleavage step. Although in vitro studies have demonstrated that two of the proteins SLPI inhibits -- elastase and cathepsin G -- can cleave gp120 (T.B. McNeely, S.P. Eisenberg, D. Dripps, and S.M. Wahl, unpublished observations), the antiviral effects of SLPI may be a consequence of binding to, or inhibition of, another as-yet-uncharacterized serine proteinase. Possibilities include tryptase TL2 (16) and CD26, a dipeptidyl dipeptidase (17), which have been suggested as the proteases responsible for cleaving the V3 loop, but SLPI does not inhibit the activity of CD26 (S.P. Eisenberg, unpublished observations), and its role in HIV infection is disputed. We are also entertaining the possibilities that SLPI interferes with the fusogenic mechanisms of the HIV envelope glycoprotein (18) or that its inhibition is the direct or indirect result of interference with viral binding or some other type of interaction SLPI may have with the target cells. One intriguing possibility revolves around earlier studies in which SLPI, administered in vivo to sheep, was found to induce glutathione (19). Some other compounds that inhibit HIV, such as N-acetyl-l-cysteine and cystamine, do so by increasing glutathione concentration (20,21), and it is conceivable that such a pathway is also triggered by SLPI. At this early juncture in defining SLPI's modus operandi, we must consider that multiple mechanisms may be involved in the antiviral activity until the emerging data force us to conclude otherwise.
Observations on SLPI in vivo are indirect or extremely sketchy at this time. Although very little is kn-own regarding the ability of SLPI to inhibit HIV in vivo, transmission of HIV via the oral cavity remains an extremely rare event, consistent with a mucosal antiviral screen that is specific to the oral cavity. SLPI is present in saliva at fairly high concentrations (~1 ug/mL), and depletion of SLPI from saliva results in a decrease in HIV-1 inhibitory action of saliva (3). In assessing in vivo localization and function of SLPI in HIV+ and HIV- individuals, we have detected similar levels of protein expression in salivary glands and in saliva of both groups. Earlier ELISA testing (6) using antibody to what was known 10 years ago as low-molecular-weight inhibitor (LMI) -- probably SLPI -- showed that the inhibitor's concentrations were high in saliva, tears, bronchial fluids, and cervical secretions; and lower in seminal fluid. Both blood serum and rectal fluid contain LMI levels as much as 1,000-fold lower than those found in other mucosal secretions -- levels that are below the minimal concentration required for in vitro antiviral activity. If these earlier data are correct, LMI (SLPI) concentrations in these various tissues may be roughly inversely related to the susceptibility to transmission for these tissues, and it is conceivable that SLPI evolved as a key protective ingredient in saliva, bronchial mucous, and cervical secretions, which coat the surfaces of the most common routes of access to the body by viruses, retroviruses, and other microorganisms. Exogenous augmentation of SLPI concentrations -- either by parenteral or topical application or through somatic gene therapy to boost expression at mucosal sites possessing subantiviral activity -- may heighten SLPI's effectiveness as a defensive shield.
At the very least, probing the SLPI barricade against HIV will help us dissect pathways of viral adherence and internalization. At best, understanding SLPI may lead to the development of new drugs that combat HIV and possibly other invasive microorganisms in a novel way.
References
1. Y. Cao, L. Qin, L. Zhang, J. Safrit, and D.D. Ho. "Virologic and immunologic characterization of long-term survivors of human immunodeficiency virus type I infection." N. Engl. J. Med. 332, 201 - 8 (1995).
2. G. Pantaleo, S. Menzo, M. Vaccarezza, C. Graziosi, O.J. Cohen, J.F. Demarest, et al. "Studies in subjects with long-term nonprogressive human immunodeficiency virus infection." N. Engl. J. Med. 332, 209 - 16 (1995).
3. T.B. McNeely, M. Dealy, D.J. Dripps, J.M. Orenstein, S.P. Eisenberg, and S.M. Wahl. "Secretory leukocyte protease inhibitor: a human saliva protein exhibiting anti-HIV-1 activity in vitro." J. Clin. Invest. (in press).
4. K. Hochstrasser, R. Reichert, S. Schwarz, and E. Werle. "Isolierung und Charakterisierung eines Proteaseninhibitors aus Menschlichem Bron-chialsekret." Hoppe Seylers Z. Phys. Chem. 353, 221 - 26 (1972).
5. H. Schiessler, M. Arnhold, K. Ohlsson, and H. Fritz. "Inhibitors of acrosin and granulocyte proteinases from human genital tract secretions." Hoppe Seylers Z. Phys. Chem. 357, 1251 - 55 (1976).
6. J.A. Kramps, C. Franken, and J.H. Dijkman. "ELISA for quantitative measurement of low molecular weight bronchial protease inhibitor in human sputum." Am. Rev. Resp. Dis. 129, 959 - 63 (1984).
7. M. Ohlsson, M. Rosengren, H. Teger, and K. Ohlsson. "Quantification of granulocyte elastase inhibitors in human mixed saliva and in pure parotid secretion." Hoppe-Seylers Z. Phys. Chem. 364, 1323 - 28 (1983).
8. R.C. Thompson and K. Ohlsson. "Isolation, properties, and complete amino acid sequence of human secretory leukocyte protease inhibitor, a potent inhibitor of leukocyte elastase." Proc. Natl. Acad. Sci. USA 83, 6692 - 96 (1986).
9. M.G. Grutter, G. Fendrich, R. Huber, and W. Bode. "The 2.5 Å X-ray crystal structure of the acid-stable proteinase inhibitor from human mucous secretions analysed in its complex with bovine a-chymotrypsin." EMBO J. 7, 345 - 51 (1988).
10. G. Stetler, M.T. Brewer, and R.C. Thompson. "Isolation and sequence of a human gene encoding a potent inhibitor of leukocyte proteases." Nucl. Acids. Res. 14, 7883 - 96 (1986).
11. S.P. Eisenberg, K.K. Hale, P. Heimdal, and R.C. Thompson. "Location of the protease-inhibitory region of secretory leukocyte protease inhibitor." J. Biol. Chem. 265, 7976 - 81 (1990).
12. Q.L. Ying, M. Kemme, and S.R. Simon. "Functions of the N-terminal domain of secretory leukoprotease inhibitor." Biochemistry 33, 5445 - 60 (1994).
13. H. Mitsuya, K.J. Weinhold, P.A. Furman, M.H. Clair, S.N. Lehrman, R.C. Gallo, et al. "3'-Azido-3'-deoxythymidine (BWA 509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T lymphotropic virus type III lymphadenopathy-associated virus in vitro." Proc. Natl. Acad. Sci. USA 82, 7096 - 7100 (1985).
14. A. Wlodawer and J.W. Erickson. "Structure-based inhibitors of HIV-1 proteases." Annu. Rev. Biochem. 62, 543 - 85 (1993).
15. A. Werner and J.A. Levy. "Human immunodeficiency virus type 1 envelope gp120 is cleaved after incubation with recombinant soluble CD4." J. Virol. 67, 2566 - 74 (1993).
16. H. Kido, A. Fukutomi, and N. Katunuma. "A novel membrane-bound serine esterase in human T4+ lymphocytes immunologically reactive with antibody inhibiting syncytia induced by HIV-1." J. Biol. Chem. 265, 21979 - 85 (1990).
17. C. Callebaut, B. Krust, E. Jacotot, and A.G. Hovanessian. "T cell activation antigen, CD26, as a cofactor for entry of HIV in CD4+ cells." Science 262, 2045 - 47 (1993).
18. O. Nussbaum, C.C. Broder, and E.A. Berger. "Fusogenic mechanisms of enveloped-virus glycoproteins analyzed by a novel recombinant vaccinia virus-based assay quantitating cell fusion-dependent reporter gene activation." J. Virol. 68, 5411 - 22 (1994).
19. A. Gillissen, P. Birrer, N.G. McElvaney, R. Buhl, C. Vogelmeier, R.F. Hoyt, et al. "Recombinant secretory leukoprotease inhibitor augments glutathione levels in lung epithelial lining fluid." J. Appl. Physiol. 75, 825 - 32 (1993).
20. M. Roederer, F.J.T. Staal, P.A. Raju, S.W. Ela, and L.A. Herzenberg. "Cytokine-stimulated human immunodeficiency virus replication is inhibited by N-acetyl-l-cysteine." Proc. Natl. Acad. Sci. USA 87, 4884 - 88 (1990).
21. T. Kalebic, A. Kinter, P. Poli, M.E. Anderson, A. Meister, and A.S. Fauci. "Suppression of human immunodeficiency virus expression in chronically infected monocytic cells by glutathione, glutathione ester, and N-acetylcysteine." Proc. Natl. Acad. Sci. USA 88, 989 - 90 (1991).