FLUORESCENCE IN SITU HYBRIDIZATION AND COMPARATIVE GENOMIC ANALYSIS
by Lance A. Liotta , M.D., Ph.D., NCI; Thomas Ried, M.D., NCHGR; and Jeffrey Trent, Ph.D., NCHGR
Fluorescence in situ hybridization (FISH) has become an increasingly
important means of identifying the chromosomal location of human genes. FISH
probes are known pieces of DNA bound to chemicals that fluoresce when excited
with a certain wavelength of light. FISH probes may be gene- or locus-specific
-- such as cosmids or yeast artificial chromosomes (YACs) -- or may be probes
that "paint" an entire chromosome (1,2). FISH is now being used in routine
prenatal screening for numerical chromosomal aberrations and diagnostic
testing for chromosomal disorders in specific diseases such as acute
lymphoblastic lymphoma. FISH is also widely used to visualize the chromosomal
locations of newly discovered genes. In this Hot Methods Clinic, we discuss the
use of FISH to localize genes, and then describe a new application of FISH --
called comparative genomic analysis -- that allows researchers to scan the
entire genome for localized or gross changes in DNA copy number.
 (16k)
(16k)
 (28k)
(28k)
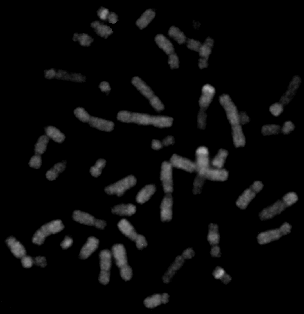
Comparative genomic hybridization (CGH) of DNA extracted from a small-cell lung cancer specimen. Only the fluorescence that is specific for tumor DNA is shown. Note the inhomogeneous staining of many chromosomal subregions, which reflects copy-number changes in the tumor genome.
The MethodAnd How It Works
Putting cDNAs On the Map
Although FISH is very useful for localizing genes within a
chromosomal band that is 10 to 20 megabases in width, it is most effective when
large insert clones are available to pinpoint the chromosomal location. At this
time, the optimal strategy for mapping a newly isolated cDNA is to combine FISH
with other genomic resources including sequence database searches and YAC
mapping. Thus, when an investigator wishes to map a new human cDNA to its
chromosomal location, genome experts such as NCHGR's Mihael Polymeropoulos
advocate first searching the sequence databases (to confirm that the gene or a
homolog hasn't already been mapped), then generating oligo probes that can be
hybridized to a series of known YAC markers that have been developed to span
the entire genome. Positive signals within this YAC pool will usually identify
a YAC contig, or collection of overlapping clones, that contains the new
marker, thus placing the cDNA on a given chromosome. If the investigator wants
to pinpoint the sequence's location more precisely, it is possible to isolate a
YAC and tag the YAC with a fluorescent marker. The labeled YAC can then be used
to perform FISH within the now-known chromosome.
As the genetic and physical map (and ultimately the sequence) for the human
genome become more complete, it is increasingly likely that mapping by computer
search will become routine, and band-region information will become available
for all markers. However, at the current time, the strategy of combining FISH
with YAC mapping is an efficient means for finding the chromosomal location of
new genes.
Comparative Genomic Hybridization
A powerful new research application of FISH, called comparative
genomic hybridization (CGH), now permits researchers to scan an entire test
genome, and in the process, highlight DNA-sequence copy-number abnormalities
on normal, reference metaphase chromosomes (3,4). Recent technical improvements
in imaging and fluorochrome application are responsible for the new FISH
refinement, which has proven especially useful in solid-tumor analysis.
A single-step CGH comparison of a normal genome with an abnormal genome can
highlight the copy-number differences for either individual whole chromosomes
or regions of specific chromosomes.
In the analysis of tumors, for example, CGH can be used to locate chromosomal
regions of amplification or deletion in paired samples of a patient's DNA,
allowing a reconstruction of the course of gene-loss or -duplication events
that have occurred in the tumor. In this instance, the investigator begins CGH
with two sets of genomic DNA: one extracted from a patient's normal tissue and
the second extracted from the tumor. Each set of DNA is labeled with a
fluorochrome of a different color. Typically, the normal genome is labeled with
a red fluorochrome, and the tumor DNA is labeled with green. Two hundred
nanograms of different sets of colored DNA are mixed and hybridized on a
metaphase spread of normal cells. The red-labeled and green-labeled sets of DNA
compete for binding to the normal chromosomes, permitting visualization through
fluorescent microscopy and computerized imaging. Each chromosome appears to be
painted with fluorescent bands of color, and the specific colors indicate
differences in the ratio of normal to tumor DNA. In chromosomal domains that
are not altered in the tumor, the red and green sets of DNA will compete
equally, resulting in a uniform hybridization pattern of red and green.
However, in areas of amplification, or increased gene copies in the tumor,
relative to the normal DNA, there will be a higher intensity of green. Areas in
which the tumor has lost DNA will be highlighted in red. Using appropriate
computerized-image analysis, the red-to-green ratio can be plotted along the
length of each chromosome, generating a survey of gross genomic changes across
all the chromosomes.
Improvements in image capture and analysis have contributed substantially to
the development of CGH. Conventional FISH is very difficult to photograph
because the images consist of pinpoint signals on a bright counterstain against
a black background. Photomicrography of these images requires long exposure
times, and the automatic exposure settings are often inaccurate because most of
the image is black.
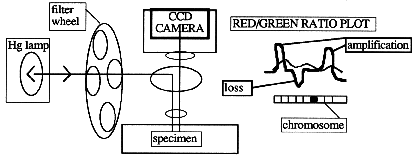
New image-capture techniques for FISH begin with the use of a cooled, digital
charge-coupled-device (CCD) camera to acquire three grayscale images (5). Each
image is captured by use of a different excitation filter, in series with a
triple-bandpass beam splitter. The filters are mounted on a wheel so that they
can be switched rapidly. The three separate images, captured with different
exposure times, are then electronically merged to form a single color image.
The entire process can be automated to produce a 24-bit color image. Operators
running commercial imaging systems can adjust the spatial resolution and
identify individual chromosomes, and software used with these systems will
generate a fluorescence red-to-green ratio along the central axis of each
chromosome. After corrections are made for uneven illumination and chromosome
overlap or bends, all 23 chromosomes can be plotted and displayed on the same
screen.
Protocol
The protocol below is taken from Trent, Thompson and Meyskens (6) and
de-scribes the preparation of metaphase chromosomes from human lymphocytes for
FISH and three-color FISH. The protocol could also be used to study cells from
other species or other human tissues, such as aneuploid tumor cells. A protocol
for Comparative Genomic Hybridization can be found in (3,4), but it involves
equipment that is not readily available. For further information on CGH,
contact Thomas Ried. Mention of a specific product does not constitute an
endorsement.
Fluorescence In Situ
Hybridization
- Sterile preparation and microdissection of metaphase
chromosomes. Using sterile technique, human lymphocyte cultures stimulated
with phytohemagglutinin (PHA) are treated with colcemid and harvested (6). The
cells are then fixed in 3:1 methanol:acetic acid for up to two hours. Next, the
metaphase cells are spread on clean coverslips (22 x 60 mm) and stored at
37oC for two to three days. Standard G-banding with trypsin-Giemsa
(GTG) (7) is performed prior to chromosomal microdisssection (2).
- Amplification of dissected DNA. Initially, cells are subjected to
eight cycles of polymerase chain reaction (PCR) (denaturation at
94oC for 1 min., annealing at 30oC for 2 min., and
extension at 37oC for 2 min.), with approximately 0.3 units of T7
DNA polymerase (Sequenase Version 2.0, USB) being added at each cycle
[Sequenase (13 units/uL) was diluted 1:8 in enzyme-dilution buffer (USB), and
0.2 uL was added to 5 uL reaction mixture] (8,9). Following this
pre-amplification step, a conventional PCR reaction, catalyzed by Taq>
DNA polymerase, is performed in the same tube. PCR reaction mix (50 uL) is then
added [10 mM Tris-HCl, pH 8.4, 2 mM MgCl2; 50 mM KCl; 0.1 mg/mL gelatin; 200 uM
each dNTP; and 2 units TaqDNA polymerase (Perkin-Elmer/Cetus)]. The
reaction mixture is heated to 95oC for 3 min., followed by 35 cycles
at 94oC for 1 min., 1 min. at 56oC, and 2 min. at
72oC, with a 5-min. final extension at 72oC.
- Fluorescence in situ hybridization. Two microliters of amplified,
microdissected DNA is labeled with biotin-16-dUTP (BMB) in a secondary PCR
reaction. This reaction is identical to the PCR reaction described above,
except for the addition of 20 uM biotin-16-dUTP. The reaction is continued for
12 to 16 cycles of 1 min. at 94oC, 1 min. at 56oC,
and 2 min. at 72oC, with a 5-min. final extension at
72oC. The PCR products are then purified through a Centricon 30
(Amicon) filter and used for FISH. Hybridization of the FISH probes follows
standard procedures (10,11). Briefly, for each hybridization, about 100 ng of
probe is used in 10 ug hybridization mixture [containing 55% formamide, 2X
standard saline citrate (SSC), and 1 ug of a DNA fraction that is enriched for
repetitive sequences, human Cot 1 DNA (Bethesda Research Laboratories)], which
is denatured at 75oC for 5 min. The slide with metaphase spreads is
denatured in 70% formamide, 2X SSC at 70oC for 2 min., and
hybridized with probes at 37oC in a moist chamber overnight. The
slide is then washed three times in 50% formamide in 2X SSC at 45oC
for 3 min. each. The hybridization signal of the probe is detected by two
layers of FITC-conjugated avidin (Vector) and amplified with one layer of
anti-avidin antibody (Vector). The slide is next counterstained with 0.5 ug/mL
propidium iodide in an anti-fade solution and examined with a microscope
equipped for epifluorescence.
- Three-color FISH. Whole-chromosome paints (WCPs) for three-color FISH
are labeled in a secondary PCR reaction identical to the one described above by
directly incorporating fluorescently tagged nucleotides. Hybridization of the
FISH probes is identical to that described above except that all three WCPs are
used simultaneously (100 ng each) in a 10-uL hybridization mixture (containing
55% formamide, 2X SSC, and 1 ug human Cot 1 DNA). Probes are detected by two
layers of FITC-conjugated avidin and amplified with one layer of anti-avidin
antibody amplified between the two avidin treatments. Slides are counterstained
with 0.5 ug/mL of the fluorescent DNA-specific dye 4,6-diamidino-2-phenylindole
(DAPI) in an antifade solution.
Contacts:
Mihael Polymeropoulos, M.D., NCHGR
phone: 402-2119
e-mail: mhp@nchgr.nih.gov
Thomas Ried, M.D., NCHGR
phone: 594-3118
e-mail: tried@nchgr.nih.gov
References
- N.P. Carter. "Cytogenetic analysis by chromosome painting." Cytometry 18, 2 - 10 (1994).
- X-Y. Guan, P. Meltzer, and J.M. Trent. "Rapid generation of whole chromosome painting probes by chromosome microdissection." Genomics
22, 101 - 7 (1994).
- A. Kallioniemi, O.P. Kallioniemi, J. Piper, M. Tanner, T. Stokke, L. Chen, et. al., "Detection and mapping of amplified DNA sequences in breast cancer by comparative genomic hybridization." Proc. Natl. Acad. Sci. USA 91, 2156 - 60 (1994).
- T. Ried, I. Petersen, H. Hotgreve-Grez, M. Speicher, E. Schrock, S. du Manoir, et. al. "Mapping of a multiple DNA gains and losses in primary small cell lung carcinomas by comparative genomic hybridization." Cancer Res. 54, 1801 - 06 (1994).
- T. Tibedo. Visualization and Analysis of FISH: Tools and Techniques. Framingham, Mass.: Vysis Inc. (1995).
- J.M. Trent, F.H. Thompson, and F.L. Meyskens. "Identification of a recurring translocation site involving chromosome 6 in human malignant melanoma." Cancer Res. 49, 420 - 3 (1989).
- R.S. Verma and A. Babu. Human Chromosomes: Principles and Techniques. New York: McGraw Hill (1994).
- S.K. Bohlander, R. Espinosa, M.M. Le Beau, J.D. Rowley and M.O. Diaz. "A method for the rapid sequence-independent amplification of microdissected chromosome material." Genomics 13, 1322 - 24 (1992).
- J. Zhang, J.M. Trent, and P.S. Meltzer. "Rapid isolation and characterization of amplified DNA by chromosome microdissection: identification of IGF1R amplification in malignant melanoma." Oncogene 8, 2827 - 31 (1993).
- P.S. Meltzer, X-Y Guan, A. Burgess, and J.M. Trent. "Rapid generation of region specific probes by chromosome microdissection and their application."
Nature Genet. 1, 24 - 8 (1992).
- D. Pinkel, J. Landegent, C. Collins, J. Fuscoe, R. Segraves, J. Lucas, et. al. "Fluorescence in situ hybridization with human chromosome specific libraries: detection of trisomy 21 and translocation of chromosome 4." Proc. National. Acad. Sci. USA 85, 9138 - 42 (1988).
Table of Contents