From Hot Starts and False Starts to Smart Starts: In Situ PCR
by Marta Corcoran , Ph.D., NCI, Mike Levin, M.D., NINDS, Steven Jacobson, Ph.D., NINDS, and Lance Liotta, M.D., Ph.D., NCI.
The enticing promise of in situ polymerase chain reaction (ISPCR) is that by amplifying DNA within cells, the sensitivity of in situ hybridization (ISH) is elevated to permit detection of single copy DNA sequences or low copy mRNAs in individual cell preparations or tissue sections. This would allow researchers to visualize single cells bearing premalignant mutations or karyotypic alterations. In addition, genetic engineers and virologists might use ISPCR to demonstrate incorporation of transfected DNA, proviral sequences or infectious agents.
How the Method Works:
The concept of ISPCR is simple. Specific nucleic acid sequences are amplified inside single cells to achieve copy numbers easily detectable by ISH. At first the procedure seems straight forward: simply place a slide containing a smear of cells or tissue section onto a thermocycler overlayed with the usual PCR reaction mixture. Following amplification, detection of product is evaluated by ISH. In practice, the simplicity is deceptive. Many NIH scientists experienced with PCR and ISH have been unable to get ISPCR to work reproducibly. For this reason, The NIH Catalyst has received a number of requests to feature ISPCR in this column.
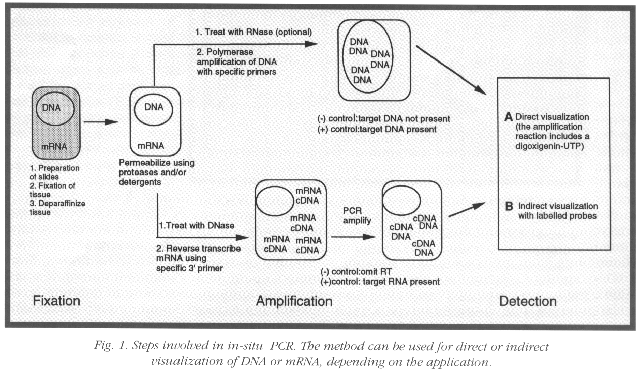
Inspecting the details of ISPCR can provide insight into the sources of artifacts. As outlined in the figure, cells or tissues are first fixed, then treated with proteinases and permeabilizing agents. This treatment allows the primers access into the cell without damaging cell morphology. Ideally, the permeabilized cells should function as "amplification sacks" (1) with semipermeable membranes allowing primers, polymerases, and nucleotides to enter the cell, but trapping the amplified target sequence inside the cell. If DNA is the target sequence, direct PCR amplification is performed. However, if the target sequence is mRNA, a preliminary reverse transcription is required to generate a cDNA template which is subsequently amplified. To confirm sensitivity and specificity, these procedures require appropriate positive and negative controls. PCR amplification is conducted in either a thermocycling oven or a block cycler. The amplified intracellular sequences are visualized directly if digoxygenin (DIG)-UTP is incorporated into the amplification reaction or indirectly by post-PCR ISH using probes that recognize sequences flanked by the PCR primers (Fig. 1).
After surveying various protocols and refinements utilized by researchers at the NIH, we are optimistic that the problems associated with ISPCR can be resolved. ISPCR can yield accurate results if the primers that yield appropriate PCR product are adapted to specific conditions so as to minimize artifacts due to diffusion of PCR products, non-specific priming and incorporation of nucleotides. A "hot start technique" for ISPCR has appeared in the literature that may reduce mispriming and primer oligomerization (2). Dr. Nuovo has published a protocol for RT in situ PCR which targets mRNA (3).
Two Protocols for in-situ PCR
Since obtaining consistent results has been challenging, we have included two different ISPCR protocols in this article. The first, by Pierre Gressens and John R. Martin of NINDS (4) directly incorporates DIG-labeled dNTP's into the amplified PCR product, and then detects the product with alkaline phosphatase. The second, by Michael Levin and Steve Jacobson of NINDS (5), utilizes overlapping multiple primer sets to retain amplified PCR products within the cell and detects the product with a specific [35]S labeled riboprobe to maximize specificity and specific activity. Mention of specific products does not constitute an endorsement.
In-Situ PCR with Direct Visualization
1. The treatment of slides, tissue fixation and de-paraffinization of tissues is similar to those used for ISH. De-paraffinize tissue using xylene and absolute ethanol followed by phosphate buffered saline (PBS) rehydration.
2. Digest the sample in 200ul of proteinase K for 5 min at 37[o]C. (For brain sections, 10, 25, or 50 ug/ml proteinase was used. For trigeminal ganglia sections, 25ug/ml of the proteinase gave the best tissue preservation for subsequent amplification of herpes simplex virus.)
3. Wash with PBS three times for 5 min. Wash 5 min with distilled water.
4. For the "hot-start" technique preheat the slides, PCR mixture, coverslips and mineral oil to 82[o]C. The PCR mixture should include the following:
0.25uM primers
10uM each dATP, dCTP, and dGTP
3.5uM dTTP
6.5uM DIG dUTP
10% glycerol
2.5 units Stoffel Taq polymerase and 2.5mM MgCl2 OR 2.5 units native Taq polymerase and 1.5mM MgCl2
5. Apply PCR mixture to slide and cover with a coverslip which could be anchored with nail polish. Blanket with a layer of mineral oil and transfer to thermocycler oven in an aluminum foil boat.
6. Amplify 1 min at 96[o]C, 1 min 59[o]C and 1min at 72[o]C. (For brain sections, amplify 15 cycles; for trigeminal ganglia use 30 cycles.)
7. Remove oil by dipping slide in xylene and absolute ethanol. Remove coverslip.
8. Wash three times for 5 min in 10mM Tris-HCl (pH 8.3) containing 1.5mM MgCl2 and 0.001% gelatin. Wash three times for 10 min in 50% formamide-2X-SSC (0.3M sodium chloride and 0.03M sodium citrate) at 37[o]C. Wash twice in 2X SSC at room temperature.
9. Detect amplified sequences by alkaline phosphatase using anti-DIG antibody as described by Boehringer- Mannheim Genius Kit 3 (Nucleic Acid Detection kit).
10. Counterstain cell nuclei and mount with aqueous mounting medium.
Protocol for In-Situ PCR with Indirect Visualization (5, 6, 8)
1. For the amplification, all solutions are made in diethyl polycarbonate (DEPC)-treated water. Fixed cell are prepared as described by Fox et al. (6). Briefly, 5 X 10[6] cells are suspended in 100-200 ul of normal human serum. A cell suspension clot is formed by adding 200-300 ul of thrombin. Clots are fixed at room temperature with 4% paraformaldehyde followed by paraffin embedding. Five micron sections of the clot are placed on silinated slides.
2. Deparaffinize slides with xylene. Rinse in absolute ethanol and rehydrate in 0.1 M Tris HCl (pH 7.4) for 10 min. Permeabilize in 0.1M Tris HCI (pH 7.4) containing 0.3% Tween 20 and 0.3% NP 40 for 10 min.
3. Digest in proteinase K (10 ug/ml) for 10 min. at 37[o]C. Wash with 0.1 M Tris/HCl (pH 7.4) three times for 5 min. each.
4. Place in prehybridization buffer (this is the "PCR buffer" without Taq, primers, or dNTP's) for 30 min. Set thermocycling oven at 72[o]C. Warm mineral oil and PCR mixture to 72[o]C. PCR buffer contains:
1 uM each primer (4 over lapping primer sets)
200 uM dNTP's
1.5 mM MgCl2
50 mM KCl
0.1 % gelatin
0.02 % NP-40
6.5 units Taq Polymerase per sample. A taq to primer ratio of 0.0125 to 0.0275 is best for PCR/ISH. The following formula is helpful to calculate the ratio. Solve X for correct amount of Taq: ratio=(X ul Taq)(5 U Taq/ul)/ (total vol in ul) (total primer concentration in pmol/ul)
5. Apply 30 ul of warmed PCR buffer to each slide and add coverslip. Dry in oven for 30 seconds to evaporate excess PCR buffer, then surround cover slip with thick layer of nail polish. Place in oven to dry for 5-6 minutes.
6. Place slide in plastic bag containing 9 ml warmed mineral oil and seal bag with heat sealer.
7. Place bag in the thermocycler oven and amplify for 40 cycles at: 92[o]C -- 1min; 53[o]C -- 1 min., 15 sec; 70[o]C -- 2 min. To maximize efficiency, oven may be placed in cold room.
8. Remove oil with chloroform and remove coverslip. Rinse again in chloroform followed by absolute ethanol for 5 min. Wash twice with PBS for 10 min.
9. Dip in 2% gelatin for 30 sec. Post-fix in 10% glutaraldehyde for 20 min and rinse in 0.3M ammonium acetate in 95% ethanol for 5 min. Air dried sections are now ready for in situ hybridization and may be stored in sealed bags with Dry-rite for several days at 4[o]C. Following PCR amplification, the ISH protocol (6) was adapted to maximize the detection of viral DNA.
10. Allow slides to equilibrate to room temperature and rehydrate in PBS for 10 min. Place slide in 1X PBS containing 10 mM dithiothreitol (DTT) 10 min. at 45[o]C. Place in 1X PBS containing 10 mM DTT, 10 mM iodoacetamide (IAA), 10 mM n-methyl maleimide (NEM) 10 min. Rinse twice in 1X PBS for 3 min.
11. Make 0.1M tri-ethanolamine hydrochloride (TEA) and raise pH to 8.0 with sodium hydroxide. Add 2.5 ml acetic anhydride to 500 ml of TEA. Drip this solution directly onto slides and rock for 10 min. Rinse in 2X SSC for 10 min.
12. Individually blot dry each slide and cover tissue section with 40 ul of prehybridization buffer for 1 hour. Prehybridization cocktail contains the following: 2X SSC, 1X Denhardt's, 50 mM phosphate buffer, 50 mM DTT, 500 ug/ul salmon sperm DNA, 250 ug/ul tRNA, 5 ug/ml poly d(A), 100 ug/ml poly (A), 0.05 pmole/ml randomer, 57% dextran/formamide (1:5=w:v).
13. In oven, denature slides at 80[o]C for 5 min and then place directly into ice-cold 2X SSC.
14. Individually blot dry each slide and add 40 ul of hybridization mixture. For the hybridization cocktail, 0.08 ul probe/ul cocktail is added ([35]S-labeled riboprobe, specific activity of 2 X 10 [6] dpm/ul). Cover with a coverslip and seal with rubber cement. Allow rubber cement to dry at room temperature for 20 to 30 min and place slides at 45[o]C overnight.
15. Remove cover slips and rinse three times in 2X SSC for 5 min. at room temperature. Incubate the slide in 0.25X SSC containing 1.2M DTT, 0.5M EDTA, and 76% deionized formamide for 30 min. at 37[o]C. Repeat incubation.
16. Rinse in 0.25X SSC containing 1.2M DTT, 0.5M EDTA, for 10 min. at 37[o]C. Place slide in RNase solution containing the following: 25 ug/ml RNase A, 25 ul RNase T1, 0.5M NaCl, 1.2M DTT in 0.01 M Tris/HCl (pH 7.4) for 40 min. at 37[o]C. Rinse in 2X SSC for 15 min. at room temp. Repeat rinse.
17. Dehydrate in 0.3M ammonium acetate in 70% ethanol for 5 min., followed by 0.3M ammonium acetate in 95% ethanol for 5 min. at room temperature and air dry.
18. Dip in NTB3 emulsion and incubate slides in dark at 4[o]C for 5 days. Develop with Kodak D-10 developer and regular fixer at 15[o]C.
19. Counter-stain with hematoxylin and eosin. Anchor coverslip with permount.
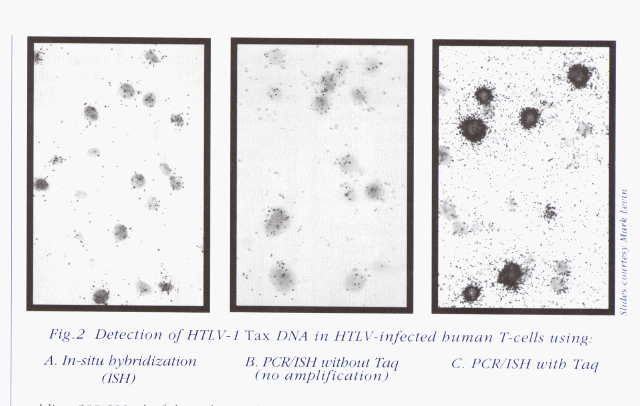
Figure 2 demonstrates how a signal obtained from ISH can be specifically amplified by using this in situ PCR technique.
Troubleshooting Tips:
"Try the conditions on a cell cytospin first before tackling an intact tissue section. If these results are negative, examine the supernatant for products to see if primers and product may have diffused out of the fixed cell. Larger products can be strategically constructed by using concatamer primers or multiple-overlapping primers (2). The use of DIG-nucleotides have also been reported to help trap the product inside the cell.
"The tissue fixation technique and chemistry must be optimized for each application. Several chemicals are available, but buffered formalin is preferred. Protease concentration, treatment time, and the method utilized for fixation must be optimized for the tissue. The goal is to permeabilize enough for the probe to penetrate cellular barriers but not destroy cellular morphology. Proteases such as pepsinogen, pepsin, or chymotrypsin could be tested at different concentrations and for different times to determine the optimal conditions for the specific application. Non-ionic detergents such as Triton-X 100 may also be used for permeabilization.
"The type of probe (RNA or DNA) and the detection method used should be optimized for each specific application. Several problems have been reported using direct incorporation of labeled nucleotides . The researcher needs to establish the best method to detect the target sequence." -- Doug Kingma, NCI.
"Perform solution-phase PCR on positive and negative areas of the tissue to confirm ISPCR results." -- John Martin and Pierre Gressens, NINDS .
"The primers to be utilized as positive and negative controls must first be tested in soluble-phase PCR to optimize the conditions for ISPCR. Aspects that need to be evaluated are: product yield, specificity of priming, optimal number of cycles, and temperature conditions for each cycle (5). According to Gressens, amplification of products of approximately 100-base pairs is best. If the probe used for the ISH was inadequate or too large, using specific primers in ISPCR may boost the strength of the signal. In-situ PCR is one of the most powerful tools for molecular biology, but it is also very tricky...Pay close attention to false positives and negatives ...Each cell should be considered a different reaction." -- Andera Cara, NCI.
In-Situ PCR Contacts
1. Mike Levin, NINDS
496-0519
2. John Martin, NINDS
496-3648
3. Andera Cara, NCI
496-8817
4. Doug Kingma, NCI
496-4969
References
1.Komminoth, P. and Long A. "In-situ polymerase chain reaction: an overview of methods, applications and limitations of a new molecular technique." Virchows Archiv. B. Cell Pathol. 64, 67 - 73 (1993).
2. G. Nouvo. "Amplifications: A forum for PCR users." Perkins Elmer Corporation 8, 1 - 5 (1992).
3. G. Nuovo "RT in situ PCR: Protocol and applications" Boehringer Mannheim's Biochemica 11, No 1 (1994).
4. P. Gressens and J.R. Martin. "In situ polymerase chain reaction: localization of HSV-2 DNA sequences in infections of the nervous system." J. Virol. Meth. 46, 61 - 83 (1994).
5. M.J. Walter, T.J. Lehky, C.H. Fox, and S. Jacobson. "In situ PCR for the detection of HTLV-I in HAM/TSP patients." Ann N.Y. Acad Sci, 724, 404 - 413 (1994).
6. C.H. Fox."In situ hybridization for detection of HIV RNA." Current Protocols in Immunology. New York: Greene publishing Assocates, 1993: 12.8.1 - 21.
7. J.F. Sallstrom, I. Zehbe, M. Alemi, and E. Wilander. "Pitfalls of in situ polymerase chain reaction (PCR) using direct incorporation of labeled nucleotides." Anticancer Res. 13 , 1153 - 1154 (19930.
8. A.A. Long, J. Mueller, J. Andre-Schwartz, K.J. Barrett, R. Schwartz, and H. Wolfe." High specificity in-situ hydridization." Diag. Mo. Pathol. 1, 45 - 57 (1992).
{kind=link}
{kind=link}