
Julie Segre
| T H E N I H C A T A L Y S T | M A Y – J U N E 2008 |
|
|
|
Another Roadmap 'Pathway'THE HUMAN MICROBIOME PROJECT
|
by Markus Elsner |
|
|
Julie Segre |
More than 90 percent of the cells in the human body belong to the bacteria, fungi, and other microorganisms that colonize our gut, skin, mouth, and other body surfaces, a fact that would probably shock most people to learn but has long been recognized by scientists.
However, notwithstanding that much is known of the diverse roles of microbial communities in a wide range of body functions—from the development of the immune system to the control of body weight and even behavior—the inventory of the kinds and numbers of microbes that live in and on the human body is surprisingly incomplete.
To remedy this knowledge gap, NIH in December 2007 launched an ambitious $115 million project to map the human microbiome, that is, to identify the species and numbers of microbes that colonize the human body as well as to characterize their protein and metabolic profile.
The NIH Human Microbiome Project (HMP), now among the “new pathways to discovery” of the NIH Roadmap, was introduced to the wider NIH community in a recent talk by Julie Segre, head of the epithelial biology section and senior investigator in the Genetics and Molecular Biology Branch, NHGRI.
Segre discussed her own pilot studies on the microbes that inhabit human skin and tied her work into the larger efforts to decipher the complete human microbiome.
Traditionally, microbiology has relied on culturing and describing individual bacterial species, but only about 1 percent of all microbes can be grown in laboratory cultures. The picture that emerges from culture-based studies, therefore, is at best semiquantitative and heavily skewed toward microbes that grow easily in the laboratory environment.
 |
 |
Janice Haney Carr, CDC,
published in the Public Health Image Library
|
Janice Haney Carr, CDC,
published in the Public Health Image Library
|
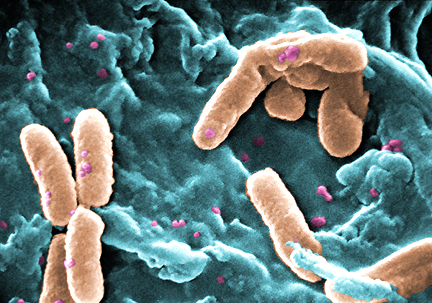
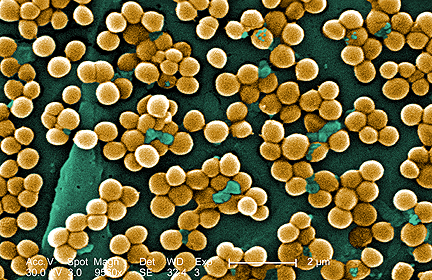
Scanning electron microscopy images of Pseudomonas aeruginosa (left) and Staphylococcus aureus, examples of the
types of bacteria found on human skin
|
|
To obtain a more balanced estimate of the bacterial population on human skin, Segre and her colleagues chose to characterize the diversity in the sequence of genes common to all prokaryotes. They selected the 16S rRNA gene, which forms a par of the bacterial ribosome that translates the genetic information into proteins. This gene is ideally suited for such an undertaking, Segre observed, because it is present in all bacteria and contains notonly regions that hardly vary between different bacterial species but also others that exhibit substantial variety in their sequences. The variable regions can be used to identify bacteria species, whereas the conserved regions are amenable to easy DNA amplification using polymerase chain reaction (PCR) techniques.
In her pilot study, Segre and her team took samples from five patients with no apparent skin disorders at the NIH Clinical Center, amplified their 16S rRNA, and determined the resulting DNA sequences. The samples were taken from the right and the left inner elbows to explore the variability of the bacterial communities in seemingly equivalent portions of the skin.
The first lesson emerging from this and other studies is that the human body is populated by representatives of only a few of the main divisions of the bacterial kingdom. Of the 70 known bacterial phyla, only four dominate the human microbiome. About 90 percent of the bacteria in Segre’s samples were members of a single phylum, the proteobac-teria—and in most patients, a single genus, the Pseudomonas bacteria, contributed about half of all microorganisms identified. [At Catalyst deadline, these studies were pending publication in Genome Research.]
How different are the bacterial communities on different parts of the skin, and how much do they vary from person to person? The samples collected from the same person showed only insignificant variation, and four of the five patients had virtually indistinguishable microbial communities. Unlike the others, the skin of the fifth patient was colonized by staphylococci, but no clinical symptoms of diseases associated with staphylococci infection were observed.
Segre compared her data from the inner-elbow samples with those collected from the inner forearm by Martin Blaser and his group at New York University and found enormous differences. While Pseudomonas dominated the inner-elbow skin, this genus of bacteria was virtually absent from the skin of the forearm, where Actinobacteria dominate.
The volunteers in both studies showed no noticeable abnormalities of the skin, and the results suggest that different parts of the skin have highly adapted and specialized groups of bacteria. Nevertheless, the number of samples collected is much too small to draw definitive conclusions, Segre observed.
An immediate question arising from these observations is whether there is a connection between the appearance of human diseases and the composition of the bacterial flora. Many human skin diseases show most of their symptoms in distinct areas of the skin. For example, atopic dermatitis, a common allergic skin hypersensitivity responsible for 10–20 percent of all visits to dermatologists in the United States, typically affects the inside of the elbow, while psoriasis, with its sore, itchy patches of thick red skin and silvery scales, which affects 2–3 percent of the population, is mainly seen on the outer elbow.
“The skin is really an ecosystem,” Segre explained. “There are niches of the skin, oily regions, moist regions, acidic regions. They all have the same human DNA . . . but may provide a different environment for different microbes to grow.”
Yet, the role of the bacterial communities in these diseases is not yet defined, but is potentially significant. In a mouse model for atopic dermatitis, Segre obtained preliminary data that the bacterial communities living on the skin of affected versus unaffected littermates are dramatically different.
Over the past few years, several research groups around the world have also used the 16S rRNA technique to characterize the bacterial communities of the mouth, colon, vagina, stomach, and esophagus. Taken together, these studies show that, depending on the location, between 20 and 80 percent of the bacterial species had not previously been described. Remarkably little had been known about even some dominant species, including the second most common genus to emerge in Segre’s study, the Janthinobacteria.
There were marked differences in the bacterial composition of the different organs—not surprising, considering the differences in their respective environments, such as the availability of nutrients, water, and oxygen.
Overall thus far, only a limited number of subjects have been sampled and only a few studies have followed the development of the microbiome over time. “What happens during puberty, for example?” asks Segre.
The first phase of the HMP aims to establish whether there is a core human microbiome. Samples from 250 volunteers will be taken at five different sites (nose, gut, vagina, skin, and mouth) and the bacterial species identified using the 16S rRNA technique. The sampling will be done at the Washington University School of Medicine in St. Louis and the Baylor College of Medicine in Houston, Texas.
The initial investigations will focus on describing the normal bacterial population of healthy human beings, the diversity within and between individuals, and the role of genetic and environmental factors in determining the composition of the bacterial communities. It will also address technical issues, such as developing methods to reproducibly take samples from the different locations.
In Segre’s study, for instance, various methods for obtaining skin samples (by punching out a section of the skin or by swapping or scraping the surface of the skin) yielded equivalent results for four of the patients, but the Staphylococcus colonization of the fifth patient would have been missed in the scraping and the punch biopsy.
 |
Jane Peterson |
Segre noted that the volunteers from rural and urban regions will be selected to encompass a wide variety of lifestyles and racial and ethnic backgrounds in order to get a glimpse of the breadth of variations that can be expected in the human microbiome. At the same time, it is unlikely that a sample of only 250 human beings will be large enough to exhaustively explore those influences. The first results from these studies are expected within 12 to 18 months.
Other questions include how many sequences are needed to obtain a complete picture or comparison of different sequencing methods.
The second phase of HMP investigations is expected to follow how differences in the microbiome correlate with lifestyle, diet, and, especially, diseases and treatment of disease in more detail. Even now, preliminary projects outside the HMP, such as the previously mentioned work on atopic dermatitis, explore the feasibility of correlative studies.
The initial phase of the HMP will contain 10 so-called demonstration projects that will explore possible connections between microbial colonization and human disease. After a year, the five most promising projects will be selected and supplied with additional scale-up funds, said Jane Peterson, associate director, NHGRI Division of Extramural Research, and HMP project leader.
Ultimately, the HMP aims to move beyond the description of species to develop a map of genes and proteins in microbial communities, describing the flow of metabolites and energy among the microorganisms and between the microorganisms and human cells. The first step in this direction is the sequencing of reference genomes of 1,000 bacterial species.
 |
Maria Giovanni |
The project started in 2007 with the awarding of $8.2 million for the sequencing of the first 200 genomes to four sequencing centers at the Baylor College of Medicine, the Washington University
School of Medicine, the Broad Institute of MIT-Harvard in Cambridge, Mass., and the J. Craig Venter Institute in Rockville, Md.
In 2009, another $30.5 million will be awarded in up to five competitive four-year grants to sequence 400 additional reference genomes and to further characterize the genetic complexity of the human microbiome. Other funding by NHGRI and international efforts will complete the full reference set of genomes.
Maria Giovanni, HMP project leader for NIAID, emphasized that the genomic information created in this part of the project will be an invaluable scientific resource comparable to the sequencing of the human genome completed in 2003.
“The scientific community will have rapid access and use the datasets and reagents generated by the HMP to ask more questions and develop research projects,” Giovanni said.
A major focus will be to develop a data coordination center to organize the large amounts of data that will be stored at the National Center for Biotechnology Information. The databases will be freely accessible to all scientists around the world.
The sequencing effort is anticipated to produce results quickly. Due to the development of new technologies triggered by the HMP and the cooperative nature of the project, Giovanni expects that the first 600–1,000 genomes will be sequenced within two to three years.