| T
H E N I H C A T A L Y
S T |
S
E P T E M B E R – O C T O
B E R 2007 |
|
 |
Iron-clad
team:
(left to right) lab chief Jeff Miller, senior author; postdoc Toshihiko
Tanno, lead author; and lab manager Terry
Lee, co-author, seated in front of computer display that summarizes
more than one million data points describing gene activity during human
erythropoiesis
|
Too
much iron will leave a body frail, destroying joints, scarring the liver, fueling
cancers and heart disease, and sapping vitality. These effects contradict everything
we have learned from the nautical sage Popeye.
The human body has no
means to shed excess iron aside from blood loss. So it regulates iron’s
uptake from food through the peptide hepcidin, which hinders iron absorption
via enterocyte cells in the gut.
The basics are well known.
In the past year, however, Jeff
Miller’s group in NIDDK’s Molecular
Medicine Branch has reached a breakthrough in understanding the underlying
chemical signaling that controls iron regulation, which could lead to diagnostic
tools and possible treatment for those with iron-overload diseases such as thalassemia.
Miller, chief of NIDDK’s
Molecular Genomics and Therapeutics Section, describes this research in the
September
2007 issue of Nature Medicine, with lead author Toshihiko
Tanno from his lab and other scientists from NIH and elsewhere.(1)
The team speculates that
the newly discovered signal, involving growth-differentiation factor 15 (GDF15),
might be common in many states of anemia and blood disorders as well as in liver
disorders and some cancers.
"While what we were
studying was a very unusual mechanism in the erythroid cells, what we may have
discovered is a very generalizable mechanism of iron regulation, or of how tissue
damage can activate certain hormones, whether it be from cancer or from liver
disease or, in our case, from anemia," Miller said. "One discovery
in a patient with thalassemia is now leading to other discoveries in other patients."
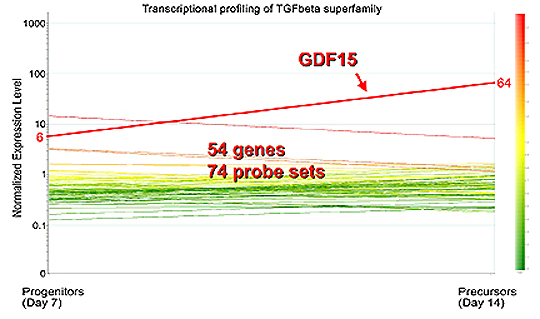 |
Identification
of GDF15 as the candidate molecule:
After transcriptional profiling of the TGF-b
superfamily members in erythroblasts, GDF15 stood out among 54 genes.
GDF15 suppresses the iron regulator hepcidin, and later it was revealed
that patients with thalassemia demonstrated 10-100–fold increases
in serum GDF15 levels when compared with healthy volunteers |
The discovery capitalized
on the unique blend of resources at NIH, such as expertise in bioinformatics,
the NIH Intramural Sequencing Center
(NISC), collaborations with smart lab neighbors, and patients at the Clinical
Center.
From
Dinner to
Bench to Bedside
Miller, a hematologist,
studies a variety of blood diseases. The core of his lab’s research since
the mid-1990s has been to map gene expression in the erythroid lineage.
"We have spent a
decade trying to put together an infrastructure for all of the genes that are
being turned on and off as an adult stem cell becomes a red [blood] cell,"
Miller said. "This information database allowed us to correlate clinical
observations with animal models and allowed us to ask what signals could possibly
regulate iron."
What hooked Miller on
iron was a conversation about a year ago at a dinner with the local chapter
of the Cooley’s Anemia Foundation, an advocacy group with members suffering
from this form of thalassemia.
Cooley’s anemia is
an autosomal-recessive blood disease affecting only about a thousand people
nationally, although single-gene carriers for thalassemia are found in 10 percent
or more of the population in Thailand, Cyprus, and other malarial regions. People
with thalassemia are unable to properly synthesize one of the globin chains
that make up hemoglobin and are thus prone to anemia. They have elevated numbers
of erythroblasts in their bone marrow but reduced numbers of healthy erythrocytes
circulating in the blood. Cooley’s
anemia and other forms of thalassemia are treated primarily by frequent blood
transfusions.
The advocacy group told
Miller its greatest concern was iron overload, a main underlying cause of death
for those with thalassemia. The excess iron can stunt growth, destroy the thyroid
gland, damage the liver, and induce diabetes, to name a few complications. Doctors
have known that, even in the absence of transfusions, thalassemia patients often
accumulate iron—and transfusions markedly worsen this.
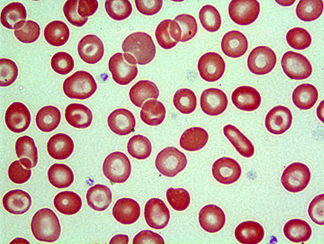 |
Bull’s
eye: Peripheral blood smear from a donor with thalassemia trait captures
classic telltale "target cells"—red cells that have a
central bull’s eye |
"Maybe there is something
about the way they make their blood—incorrectly—that makes them absorb
too much iron," Miller hypothesized.
Miller had hints from
several sources—including the clinic he established at the Clinical Center
and an animal model from the neighboring lab of Chuxia
Deng, chief of the NIDDK Mammalian Genetics Section—that the signal
could be a member of the transforming growth factor–b
(TGF-b) superfamily of genes. He was particularly
inspired by Deng’s work, which he had heard at a NIDDK retreat, and began
to concentrate on TGF-b proteins, including GDF and
bone morphogenetic proteins, all in the superfamily. He was also inspired to
pursue the project by Carolyn
Philpott, senior investigator in NIDDK’s Liver
Diseases Branch and an expert on iron biology.
Alan
Schechter, the NIDDK Molecular
Medicine Branch chief, described Miller’s lab’s process of narrowing
in on the suspected genes as going from "bedside to bench to bedside,"
one of the true advantages of the intramural research program.
"Jeff established
two years ago a clinic in the hospital to see patients," Schechter said.
"Once you have the robust clinical base, then you can go back and forth.
. . . It’s an iterative process. You can’t just think some profound
thought in a laboratory and then come over to the hospital in the middle of
the night to apply it to patients."
Erythroid
Gene Clearinghouse
 |
Susan
Leitman |
 |
Gerard
Bouffard |
 |
Alan
Schechter |
Tanno,
an IRTA postdoc fellow, began the project with the meticulous process of identifying
all the signals made by adult stem cells during their transformation from erythroblasts
to red blood cells. Key to this work was the warehouse of data from the Human
Genome Project and bioinformatics tools to analyze these data. A variety of
bioinformatic approaches was used, including those available through NCBI,
commercial software for array analyses, and custom programming strategies crafted
by Gerard Bouffard of NHGRI, director
of the NISC Bioinformatics Group.
The process entailed isolating
mRNA from erythroid precursor cells, reverse-transcribing this to cDNA, creating
thousands of expressed sequence tags (ESTs), removing contaminants, and then
"BLASTing" them using NCBI’s BLAST
tools to compare them with other gene sequences.
"BLAST is one of
those tools you can’t imagine modern life without," said Bouffard,
who wrote a complementary software program for Miller’s group "to
distill out gold nuggets" from the BLAST results. He has worked with Miller
on this project for several years.
All this information is
posted at NIDDK’s Hembase,
a public repository
of information on erythroid ESTs and also sequences encoding several hundred
additional genes with known expression in erythroid cells, organized and linked
according to the location of these sequences within the human genome.
From
Hint to Confirmation
Guiding Tanno on his needle-in-a-haystack
search for an iron regulator in the midst of all these sequences was the clinic-
and lab-driven hypothesis that iron regulation involved the TGF-b
superfamily. He examined adult human erythroid cells from clinic patients to
see whether any TGF-b genes were expressed.
Three candidates emerged
from a long list, with GDF15 in particular copiously expressed by erythroid
cells.
Going back to the clinic,
the team checked whether patients with thal-assemia had too much GDF15 in their
blood. The answer was as clear as a bell.
"They had incredibly
high levels," Miller said, "a logarithmic increase in the level of
this in their blood" relative to the patient’s iron overload and compared
with healthy patients. "We were thinking, wow. . . . It was very satisfying
to be able to go back to the clinic to these patients who were donating their
blood to figure out if our hints in the laboratory were useful."
Once the group found what
was increased in the blood, they could determine the mechanism: GDF15 appears
to be an inhibitor of hepcidin, a major iron regulator made in the liver. This
hepcidin suppression leads to more iron absorption.
"Since erythroblasts
need iron to make hemoglobin, we reasoned that the increased number of erythroblasts
in thalassemia may send stronger messages to the liver to suppress hepcidin
and thereby absorb more iron even in the condition of iron overload," said
Tanno.
Broader
Implications
Miller has about a dozen
thalassemia patients in his clinics, but he has been able to strengthen his
ongoing research on this topic by studying blood from other patients, including
those with other iron-overload diseases seen by Susan
Leitman, chief of the Blood
Services Section of the CC’s Department
of Transfusion Medicine, as well as by receiving samples from thalassemia
patients in Thailand, where nearly 1 percent of the population has some thalassemia
disease.
"Many experts in
iron regulation have postulated the existence of an ‘erythroid regulator’
of iron absorption for a while now,"said Leitman. "The search for
this erythroid regulator has been a hot issue in labs focused on iron homeostasis.
Jeff’s identification of GDF15 is a major breakthrough in this field."
The Thai samples came
courtesy of Suthat Fucharoen of Mahidol University in Nakornpathom, Thailand,
who had spent a summer at NIH several years ago and who has continued to collaborate
with Miller, Schechter, and NIDDK Director Griffin
Rodgers.
"This is the basis
of how science works—exchanging visits and established collaborations,"
Schechter said, adding that Fucharoen’s collaboration has greatly aided
in the discovery of iron regulation.
There are other anemias
that don’t behave like thalassemia but still might be regulated by hepcidin
and GDF15, Miller said. He also speculates that the involvement of GDF15 in
thalassemia and other diseases, including cancers, may extend beyond the regulation
of hepcidin.
"We’re hoping
now—very much so—we can use this for diagnostic, prognostic, and
even therapeutic purposes, because the discovery of signals immediately leads
to the possibility of blocking those signals," Miller said.
"Information that
has come from the study of hemoglobin diseases has been applicable to lots of
other diseases," said Schechter, citing Linus Pauling’s 1949 paper
defining sickle cell as the first known molecular disease. What Miller has done,
Schechter added, was make "use of the new era of genomic medicine to define
a subfield that probably could be called human erythroid cell genomics . . .
to pioneer a whole new field within hematology."
Whereas the current project
has been focused almost entirely on thalassemia, the lab hopes to apply this
translational approach to other diseases and to ensure that the broader scientific
community has access to their genomic studies to facilitate basic and clinical
connections that would otherwise be difficult to make.
"All this information
can now be organized because of the Human Genome Project. Let’s not forget
that," Miller said. "Now we’re taking the next step—and
perhaps the more difficult though most important step—to use the information
to make people feel better." 
1. T.
Tanno, N.V. Bhanu, P.A. Oneal, S.-H. Goh, P. Staker, Y.T. Lee, et al., "High
levels of GDF15 in thalassemia suppress expression of the iron regulatory protein
hepcidin," Nature Medicine 13, 1096–1101 (2007); published
online, August 26, 2007.
Return
to Table of Contents