
Rémy
Bosselut
| T H E N I H C A T A L Y S T | M A Y – J U N E 2007 |
|
|
|
| P E O P L E |
RECENTLY TENURED
|
|
Rémy
Bosselut
|
Rémy Bosselut trained at the Institut Curie in Orsay, France. He earned his M.D. degree in 1992 from the Xavier Bichat School of Medicine and his Ph.D. degree in 1993 from the University Denis Diderot, both in Paris. After postdoctoral training at the NCI Experimental Immunology Branch with Al Singer, he joined the Laboratory of Immune Cell Biology as a tenure-track investigator in 2000 and is currently a senior investigator in that lab.
Our laboratory investigates the transcriptional control of T-lymphocyte differentiation. A critical component of the immune system, T cells derive from bone marrow precursors that undergo extensive proliferation and differentiation in the thymus. The two main T-cell subsets have notably different functions: Those that express the CD4 surface molecule control the function of most other immunocompetent cells, and those that express the CD8 surface molecule hunt and destroy cells infected by intracellular pathogens. These subsets actually constitute two separate lineages that diverge in the thymus from a common precursor, the so-called double positive (DP) that expresses both CD4 and CD8.
Our work focuses on the differentiation in the thymus of mature T cells from immature precursors (thymocytes)—and, specifically, on how DP thymocytes choose either the CD4 or CD8 lineage.
Our current approach to this question began a few years ago with microarray gene expression analyses that led us to identify a zinc-finger transcription factor, known as cKrox or Thpok, as a major CD4-differentiating factor.
We showed, first, that this gene is expressed in CD4- but not CD8-lineage cells and, second, that enforcing cKrox expression in DP thymocytes prevents their differentiation into CD8 cells. The cells that would normally become CD8 adopt a CD4 fate; that is, cKrox both inhibits the CD8- and promotes the CD4-differentiation program.
Our current research focuses on these questions:
![]() How is cKrox expression controlled in thymocytes and, especially, how is it
confined to the precursors that will eventually become CD4 T cells?
How is cKrox expression controlled in thymocytes and, especially, how is it
confined to the precursors that will eventually become CD4 T cells?
![]() How does cKrox promote CD4 differentiation in the thymus? What are its target
genes? Does it interfere with other transcription factors involved in CD4–CD8
differentiation and, if so, how?
How does cKrox promote CD4 differentiation in the thymus? What are its target
genes? Does it interfere with other transcription factors involved in CD4–CD8
differentiation and, if so, how?
![]() Our third area of investigation derives from experiments in which we forced
the expression of cKrox in mature CD8 T cells (in which it is normally not expressed).
We found that cKrox downregulates expression of genes characteristic of CD8
differentiation, including those encoding CD8 molecules themselves or cytotoxic
proteins such as perforin or granzyme B. These findings indicated that that
these fully differentiated CD8 T cells remained sensitive to the repression
by cKrox of the CD8 differentiation program. However, unlike in thymocytes in
which cKrox both prevents CD8 and promotes CD4 differentiation, expression of
cKrox in differentiated CD8 cells failed to revert them to the CD4 lineage.
Our third area of investigation derives from experiments in which we forced
the expression of cKrox in mature CD8 T cells (in which it is normally not expressed).
We found that cKrox downregulates expression of genes characteristic of CD8
differentiation, including those encoding CD8 molecules themselves or cytotoxic
proteins such as perforin or granzyme B. These findings indicated that that
these fully differentiated CD8 T cells remained sensitive to the repression
by cKrox of the CD8 differentiation program. However, unlike in thymocytes in
which cKrox both prevents CD8 and promotes CD4 differentiation, expression of
cKrox in differentiated CD8 cells failed to revert them to the CD4 lineage.
We are currently investigating the bases for this difference and notably whether epigenetic marking of CD4-lineage genes in CD8 cells would prevent their reactivation despite cKrox expression.
We expect that these experiments will shed light on the mechanism of CD4-CD8 differentiation in the thymus, a process crucial to the proper functioning of the immune system. From a broader point of view, these analyses should contribute to deciphering the mechanisms by which lineage fate is determined and maintained during mammalian development.
 |
|
Richard
Childs
|
Richard Childs received his medical degree from Georgetown University in Washington, D.C., in 1991. He completed his internship and residency in internal medicine at the University of Florida in Gainesville and served as the chief medical resident there from 1994 to 1995 before coming to NIH in 1995. He completed a fellowship in medical oncology at NCI and then trained as a hematology fellow at the Hematology Branch, NHLBI, where he conducted research on allogeneic bone-marrow transplantation in the laboratory of John Barrett. He became a tenure-track investigator in 1999 and is currently a senior clinical investigator in that branch and a commander in the United States Public Health Service.
My research interests have focused on tumor immunology and allogeneic immunotherapy for solid tumors and hematologic malignancies. Research conducted during my hematology fellowship showed that metastatic solid tumors could be killed by allogeneic T cells.
My clinical research program has focused on expanding the application of nonmyeloablative allogeneic hematopoietic stem-cell transplantation to treat solid tumors and nonmalignant hematological disorders such as paroxysmal nocturnal hemoglobinuria (PNH).
We demonstrated that PNH, a debilitating disorder of hematopoietic stem cells associated with bone marrow failure, red blood cell hemolysis, and blood clot formation, could be cured immunologically by transplanted donor T-cells that eradicated PNH stem cells.
Since 1999, more than 225 patients have undergone allogeneic hematopoietic stem-cell transplantation on our protocols. These clinical research trials advanced the field of hematopoietic stem-cell transplantation, providing new insights into the full therapeutic potential of allogeneic immunotherapy to treat both nonmalignant hematological disorders and metastatic kidney cancer.
Our publication in the New England Journal of Medicine in 2000 showed that renal cell cancer could be eradicated in patients with treatment-refractory metastatic disease after nonmyeloablative stem-cell transplantation, which spawned the development of allogeneic transplant trials nationally and internationally for a variety of solid tumors.
In addition to our clinical research program, my laboratory conducts basic and translational research in the field of tumor immunology. We are currently focusing on tumor antigen discovery and methods to enhance graft-vs.-tumor (GvT) responses against cancer using natural killer (NK) cells.
Using human T cells obtained from kidney-cancer patients whose tumors regressed after nonmyeloablative transplantation, my group recently identified a novel gene derived from a human endogenous retrovirus (HERV) whose expression appears restricted to clear-cell renal-cell carcinoma.
Importantly, more than 50 percent of kidney cancers express this gene, and peptide antigens derived from this HERV are immunogenic, eliciting a tumor-specific T-cell response against kidney cancer. We are actively investigating the function of HERV in kidney cancer and the development of a tumor vaccine targeting HERV-derived antigens.
We have also focused on methods to enhance the antitumor effects of both allogeneic and autologous NK cells. Through in vitro experiments, we discovered that allogeneic NK cells with KIR (killer inhibitory receptor) incompatibility have enhanced antitumor activity against kidney cancer and melanoma compared with autologous NK cells.
When adoptively infused in tumor-bearing mice that have undergone an allogeneic transplant, these NK cells reduce graft-vs.-host disease (GvHD) and mediate potentially curative graft-vs.-solid tumor effects. Based on these findings, my group is designing a clinical trial to test the ability of in vitro expanded and adoptively infused allogeneic NK cells to prevent GvHD in patients with PNH and aplastic anemia and to enhance GvT effects in patients with malignancies who are undergoing hematopoietic stem-cell transplantation.
In our exploration of ways to potentiate the antitumor effects of autologous NK cells isolated from cancer patients, we discovered that tumors exposed to the proteosome inhibitor bortezomib and the histone-deacetylase inhibitor depsipeptide are more vulnerable to autologous NK effects. Both drugs were found to upregulate death receptors on the tumor surface for ligands expressed on NK cells. Furthermore, in a tumor-bearing mouse model, we found that pretreatment with bortezomib enhanced the antitumor activity of adoptive infusions of autologous NK cells.
Based on these findings, our group received a "bench-to-bedside" award in 2006 to evaluate the potential of bortezomib to enhance the antitumor effects of adoptively infused NK cells in humans with variety of metastatic cancers. A scale and validation testing of this approach is currently being performed in the CC Department of Transfusion Medicine. We anticipate enrollment of patients in this trial within the next three to six months.
 |
|
Ira
Daar
|
Ira Daar received his Ph.D. from the State University of New York at Buffalo in 1988 and came to NIH for postdoctoral training with George Vande Woude in the ABL/Basic Research Program. In 1992 he became a senior staff fellow and in 1997 a principal investigator in the Regulation of Cell Growth Laboratory and the Laboratory of Protein Dynamics and Signaling. He is currently head of the Developmental Signal Transduction Section and a senior investigator in the Laboratory of Cell and Developmental Signaling, NCI.
My long-standing interest is to understand the basic signaling pathways that regulate morphogenetic events during development. These pathways control cell movement and tissue reorganization during early embryogenesis and, when deregulated, may contribute to human disease.
The Xenopus model system has proven to be a tractable one for uncovering signaling pathways that regulate cell cycle progression and cell movement. My primary focus is on members of the Eph family of receptor tyrosine kinases and their membrane-bound ligands, the ephrins. The Eph/ephrin signaling pathway plays an important role in controlling the movement and positioning of cells during development.
Eph receptors and ephrins are also overexpressed in a wide variety of human tumors and cancer cell lines, with a particularly high frequency of deregulation observed in metastatic tumor cells. Moreover, these proteins have been implicated in tumor-cell invasion and tumor angiogenesis.
Therefore, further elucidating the precise mechanisms and proteins involved in Eph/ephrin signaling is likely to provide valuable insight into cancer dynamics as well as into normal morphogenetic processes.
My laboratory has explored a new concept for the signal-transduction cascade initiated by ligand-receptor interactions known as "bidirectional signaling," in which a transmembrane ephrin ligand not only activates a cognate Eph receptor on a neighboring cell, but also transduces a signal through its own intracellular domain. These studies have revealed that signaling from ephrinB1 affects cell adhesion and movement.
Further, my laboratory has shown that there is signaling cross-talk between the Eph and FGF receptor (FGFR) tyrosine kinase pathways. Through biochemical studies, we have found that activated FGFRs can interact with and regulate the signaling properties of ephrinB1 via tyrosine phosphorylation of the ephrinB1 intracellular domain.
In collaboration with Sally Moody, of George Washington University in Washington, D.C., and Kathy Moore, of the University of Utah, Salt Lake City, we have been using the Xenopus eye field as a model system. We have been able to demonstrate the functional consequence of FGFR/ephrinB1 signaling cross-talk in the regulation of stem-cell movement and cell fate. This work linked these two key signaling pathways (Eph and FGFR) as co-regulators of an important morphogenetic process and provided evidence that alterations of cell fate can result from changes in cell movement.
Based on these findings, my colleagues and I have proposed a model in which the appropriate in vivo positioning of cells occurs as a result of the expression patterns and interactions between ephrins and their cognate receptors and alternative growth factors.
My laboratory has continued to take advantage of the eye-field system, and our most recent studies have revealed that ephrinB1 controls retinal progenitor cell movement by interacting with the Dishevelled (Dsh) adaptor protein and co-opting downstream members of the noncanonical Wnt/planar cell polarity (PCP) pathway.
Importantly, these studies connect ephrin signaling with the PCP pathway and have set the stage for my future studies examining the mechanism by which ephrinB1 influences this pathway.
We now have evidence that ephrinB1 interacts with the Par polarity complex, which is a major determinant in establishing tight junctions. Through this interaction ephrinB1 can exert regulatory control over tight-junction assembly. Using in vivo approaches, we can now mechanistically test how ephrinB1 regulates cell-cell adhesion during development.
We will continue studies to broaden our concept and knowledge of signaling pathways that regulate cell movement and cell polarity during embryogenesis, with the idea that this knowledge may also affect how we approach therapeutic applications for metastatic disease.
 |
|
Daniel
Douek
|
Daniel Douek received his medical degree from the Universities of Oxford and London in 1990 and then his Ph.D. in immunology from the University of London in 1996. He did his postdoctoral training with Richard Koup at the Rockefeller University in New York and at the University of Texas Southwestern Medical Center at Dallas and became a tenure-track investigator at the Vaccine Research Center, NIAID, in 2000. He is currently a senior investigator, leading the Human Immunology Section in the Laboratory of Immunology, VRC.
We in the Human Immunology Section study the processes that determine the course of human diseases in which the immune system, particularly its T-cell arm, plays a central role in pathogenesis and outcome. We aim to use the knowledge gained to initiate clinical studies of new therapeutic and vaccine approaches.
Our overriding strategy is to address questions of human disease directly in humans and nonhuman primates, with as little in vitro manipulation as possible and with an emphasis on elucidating the basic mechanisms that underlie disease processes.
We are studying the nature and dynamic interactions of HIV-specific T-cell clones in HIV disease and after vaccination against HIV; our goal is to establish correlates of effective and protective immunity.
Such findings will in turn open lines of further inquiry for researchers in developing strategies using T-cell immunity to fight HIV infection.
Specifically, our recent studies have shown that the use of HIV-specific CD8 T-cell clones with particular T-cell receptors (TCR) may affect biological outcome in infected individuals. We aim to understand these phenomena at the level of TCR affinity, precursor frequency, cross-reactivity, and structure.
By studying the mechanisms underlying HIV pathogenesis and immune reconstitution after therapy, we aim to understand the tempo and mechanism of HIV disease progression and how recovery from HIV disease can be enhanced.
The main thrust of our work at the moment is to investigate CD4 T-cell depletion at mucosal sites, the consequences of damage to the gut mucosa, and how such damage underlies disease progression to AIDS. We recently reported that microbial translocation from the gut lumen into the systemic circulation causes immune activation and may contribute to progressive disease.
We hope that these approaches contribute to both prevention and treatment of HIV disease in humans.
 |
|
Stewart
Levine
|
Stewart Levine received his M.D. from the State University of New York School of Medicine, Stony Brook, in 1983. He completed an internship and residency at St. Vincent’s Hospital and Medical Center in New York City and a fellowship in pulmonary medicine at the Memorial Sloan-Kettering Cancer Center before coming to NIH in 1989 for clinical and postdoctoral training in the CC Critical Care Medicine Department. He subsequently became a staff clinician in that department. In 1998, he joined the Pulmonary-Critical Care Medicine Branch, NHLBI, as a tenure-track investigator and is now a senior investigator and head of the section on lung inflammation in that branch.
Our research has focused on identifying molecular mechanisms by which inflammation can be regulated. Because of the important role of tumor necrosis factor (TNF) in inflammatory lung diseases and critical illness, we have studied the type I, 55-kDa TNFR1 (TNFRSF1A) as a model system. One mechanism by which TNF bioactivity is regulated involves the shedding of cell surface receptors, which then function as soluble TNF-binding proteins. Prior work in the field established that soluble TNF receptors are generated by the proteolytic cleavage of receptor ectodomains by a sheddase, such as TNF-a converting enzyme (TACE), also known as ADAM17.
Our studies have identified new molecular mechanisms regulating the release of TNFR1 to the extracellular space. We discovered that a full-length TNFR1 is constitutively released from human vascular endothelial cells within the membranes of exosome-like vesicles.
This release of TNFR1 exosome-like vesicles represents a novel alternative pathway for the generation of soluble cytokine receptors that is distinct from the proteolytic cleavage of receptor ectodomains or the translation of an alternatively spliced mRNA. We confirmed the in vivo significance of these observations in clinical studies, which demonstrated TNFR1 exosome-like vesicles in human plasma and bronchoalveolar lavage fluid.
To better understand the intracellular pathways that modulate soluble TNFR1 generation, our laboratory identified, cloned, and characterized ARTS-1 (aminopeptidase regulator of TNF receptor shedding) as a type II integral membrane zinc metalloprotease. ARTS-1 is a TNFR1-binding protein that regulates both the constitutive release of TNFR1 exosome-like vesicles and the IL-1b–mediated inducible proteolytic cleavage of TNFR1 ectodomains, but does not appear to function as a TNFR1 sheddase. We also showed that ARTS-1 regulates the release of the soluble cleaved forms of IL-6Ra and IL-1RII.
Therefore, ARTS-1 regulates the release of three distinct cytokine receptor superfamilies: those of the TNF receptor superfamily (TNFR1), the class I cytokine receptor superfamily (IL-6Ra), and the immunoglobulin/Toll-like receptor superfamily (IL-1RII).
Our laboratory subsequently identified nucleobindin 2 (NUCB2, NEFA) as a calcium-dependent, ARTS-1–binding protein that associates with intracytoplasmic TNFR1 vesicles. We showed that the calcium-dependent formation of ARTS-1/NUCB2 complexes are required both for the constitutive release of TNFR1 exosome-like vesicles and for the IL-1b–mediated, inducible proteolytic cleavage of TNFR1.
Therefore, NUCB2 and ARTS-1 regulate two zinc metalloprotease–dependent mechanisms of cytokine receptor shedding: the sheddase-independent constitutive release of exosome-like vesicles containing full-length TNFR1 receptors and the sheddase-dependent inducible proteolytic cleavage of receptor ectodomains.
In collaboration with Martha Vaughan and Joel Moss in our branch, we recently identified a vesicular trafficking pathway that mediates the release of TNFR1 exosome-like vesicles. We showed that the brefeldin A–inhibited guanine nucleotide-exchange protein BIG2 associates with TNFR1 and selectively modulates the constitutive release of TNFR1 exosome-like vesicles via an ARF1- and ARF3-dependent mechanism. This functional interaction between TNFR1 and BIG2 is downstream from the ARTS-1/NUCB2 complexes.
Our research program will remain focused on investigating regulatory mechanisms that modulate inflammation. In particular, we are continuing our studies to identify and characterize the molecular pathways that regulate the release of soluble cytokine receptors that can modify inflammatory and immune events.
We are also planning clinical and translational studies to identify new therapeutic approaches for patients with asthma, especially those with severe disease that cannot be adequately controlled with current medications.
 |
|
Ian
Macdonald
|
Ian Macdonald received his M.D. degree from McGill University in Montreal in 1979. He did his postdoctoral training at the University of Ottawa and became professor and chairman of the Department of Ophthalmology at the University of Alberta in Edmonton before joining NIH in 2007 as chief of the Ophthalmic Genetics and Visual Function Branch, NEI.
Medical genetics is about people and journeys in time. During my undergraduate days at McGill University, I was introduced to genetics by Clarke Fraser and Charles Scriver, who were keenly interested in the genetics of human dysmorphology and human metabolic conditions. That experience set the mold for a lifetime interest in genetics and led me to a career in medical genetics.
I was introduced to vision research early in my fellowship training in clinical genetics with Alasdair Hunter at the University of Ottawa: A patient asked if I would do research on her family, many members of which were affected by a degenerative retinopathy called choroideremia (CHM). I was just learning to use restriction-fragment-length polymorphisms as markers to map a genetic locus. CHM was X-linked, and there were few markers beyond Xg and color-blindness that one could use to determine linkage.
Molecular markers offered the potential to link the eye disorder to a specific arm of the X chromosome and, with luck, to get close enough to make some guesses as to where the gene was.
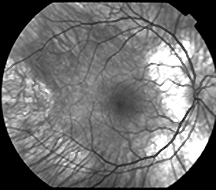 |
|
Fundus
of a male patient with choroideremia
|
I was successful in linking the disorder to the long arm of X. At that time, single-nucleotide polymorphisms did not exist and there were few other markers available to refine the map location. Having exhausted genomic markers, I began with a then-graduate student, Paul Wong, to isolate expressed sequences from the X chromosome that might be part of candidate genes for CHM. In the early 1990s, I began to characterize mutations in the choroideremia gene (CHM) in families originally involved in mapping. That activity enabled what became a reference laboratory for this condition, funded by the Foundation Fighting Blindness.
The CHM gene product, Rab escort protein–1 (REP-1), has an important role to play in the trafficking of vesicles involved in intracellular transport in all cells—in particular, trafficking within the eye. In collaboration with NEI’s Bob Fariss and Chi-Chao Chan, I have shown that the REP-1 protein is expressed abundantly in the eye. This work, as well as my continuing study of CHM gene expression in the eye, has promising implications for future therapeutic approaches.
For the past 15 years, I have studied genetic isolates from people living in the province of Alberta, where heritable ocular conditions are prevalent. Our early mapping studies in a family with autosomal dominant form of Stargardt-like macular dystrophy lead to the isolation, with collaborators, of ELOVL4, the gene that is mutated in this condition. This gene is thought to be involved in the pathway of de novo synthesis of very-long-chain polyunsaturated fatty acids, including docosahexaenoic acid (DHA). DHA is the major fatty acid of the retina and is essential for normal retinal development and physiology.
I have completed two pilot clinical studies on the effect of DHA as a nutritional supplement on macular function in patients with this form of Stargardt disorder. The resuts have been encouraging, and a clinical trial of DHA supplementation in patients with the dominant form of Stargardt-like macular dystrophy is planned to begin at NIH.
Meanwhile, because launching clinical trials in families with few affected members can be challenging, and animal studies can be helpful in suggesting clinical study design and potential outcomes, I am also continuing to work with animal models with colleagues at the University of Alberta—Yves Sauvé and Silvina Mema. We are carefully studying the effect of an elovl4 mutation on the retina and on measurable functional changes in these mutant mice as they age, as well as whether a high-DHA diet will alter their retinal function.
 |
|
Ruth
Pfeiffer
|
Ruth Pfeiffer trained in applied mathematics at the Technical University of Vienna and received a Fulbright Fellowship to study applied statistics. She earned a Ph.D. in mathematical statistics from the University of Maryland, College Park, in 1998, and joined NIH as a Cancer Research Training Award fellow in the Division of Cancer Epidemiology and Genetics (DCEG), NCI. She was promoted to research fellow in October 2000, and, since 2001, has been a principal investigator in the Biostatistics Branch.
In recent years, I have initiated methodologic projects in several areas, including latent class models for viral epidemiologic and laboratory studies, design of genetic epidemiologic studies, models for family-based association studies, and statistical methods for risk projection and classification.
A focus of DCEG is to clarify the relationship of infectious agents, especially viruses, to human cancer. I have been involved in several studies focused on the biology, transmission routes, and natural history of human herpesvirus 8 (HHV-8), also known as KSHV, the infectious cause of Kaposi’s sarcoma.
I developed a Markov model to estimate the risk of transmission of HHV-8 from transfusion in Ugandan children with sickle cell anemia. The analysis was based on cross-sectional age-specific seroprevalence data and on the individuals’ transfusion histories; it estimated that the risk of transmission of HHV-8 from blood transfusions was about 2 percent.
This model was crucial to the study because standard statistical approaches would not have estimated the critical quantity of interest, namely, the per-transfusion risk of infection with HHV-8. This analysis also allowed investigators to compare transfusion risk with background sources of risk.
Another challenge is estimation of HHV-8 prevalence. Although infection status with HHV-8 can be assessed by several serological assays, there is no gold-standard measure for a definitive diagnosis. To address this issue, I developed and applied mixture models to estimate the prevalence of infection of agents for which there is no definitive assay.
The mixture approach assumes that the observed assay distribution in a population is generated by different distributions for infected and uninfected subjects; this assumption allows one to determine the fraction of the population that is infected. I have used this approach to estimate the prevalence of HHV-8 in several African populations.
DCEG is leading many studies of genetic contributors to cancer etiology. Assessing whether disease aggregates in families is a first step in identifying a genetic component of that disease. I have adapted survival methods to test for familial aggregation of disease in cancer registry data that define familial relationships and identify all diseased cases in a population. I used these methods in collaborative studies of familial aggregation of lymphoproliferative disorders in Denmark and Sweden.
I also developed multilevel random-effects models that were used to analyze the effects of covariates in family-based case-control studies in which families are ascertained only if they have several affected members. This model accounts for random family effects as well as random genetic effects that induce specialized forms of correlation within families. Because these families are specially ascertained, methods for simple random samples of families do not apply, and special computational methods are needed.
The candidate-gene approach to studying genetic associations with disease is based on investigating several single-nucleotide polymorphisms (SNPs) in a gene that may be associated with disease. I have assessed sample-size requirements and power for population-based and family-based case-control studies to detect associations between disease and a genetic marker, instead of the true disease gene locus. Such calculations are crucial in designing association studies.
I am also interested in risk prediction, discrimination, and related issues and have developed an absolute risk–prediction model for colorectal cancer, the second leading cause of cancer death in the United States. The absolute risk is the probability that an individual with given risk factors and a given age will develop colorectal cancer over a defined period of time.
In collaboration with my branch chief, Mitchell Gail, I defined loss-function–based criteria tailored to specific applications to evaluate models of absolute disease risk. This approach showed that models with modest discriminatory power may not be useful for screening but may be useful in making clinical decisions that involve potential for both benefit and harm.
I have also developed classification algorithms for high-dimensional data—for example, microarrays, mass-spectrometry data, and denaturing high-performance liquid-chromatography curves. With my fellow Annette Molinaro, now at Yale, I compared cross-validation methods used to estimate prediction errors for various discrimination algorithms based on high-dimensional data.
All of my work has been directly or indirectly motivated by collaboration with my colleagues at the DCEG and elsewhere at NIH. My training in mathematics and the scientific environment of DCEG have yielded a rich and enjoyable statistical and collaborative research program for epidemiological and laboratory studies.
 |
|
Ying
Zhang
|
Ying Zhang received her Ph.D. from the University of Wisconsin–Madison in 1995, carried out her postdoctoral training with Rik Derynck at the University of California, San Francisco, and joined the Laboratory of Cellular and Molecular Biology , NCI, in 2000 as a tenure-track investigator. She is currently a senior investigator in that lab.
The research focus of my laboratory is on the roles and signaling mechanisms of the TGF-b superfamily of peptide growth factors that perform a diverse array of functions during development and cancer.
My interest in the TGF-b pathway began during my postdoc training at UCSF, where I was among the first few scientists to identify and clone the mammalian Smad proteins that later turned out to be the substrates of TGF-b receptor kinases and the major intracellular signaling mediators for all members of this family. My work there also helped to define the mechanism of Smad3 in cooperatively interacting with c-Jun/Fos to control TGF-b target gene expression, which is still today is the paradigm for how all Smads function in transcription.
After joining the Laboratory of Cellular and Molecular Biology at NCI, I directed the effort of my group to three unresolved issues in the TGF-b pathway—the nature and significance of a Smad-independent TGF-b receptor-signaling mechanism, the physiological functions of two newly isolated ubiquitin E3 ligases for Smad proteins, and the role of Smad3 in tumorigenesis.
Based on the crystal structural data and kinase-substrate interaction, we constructed a mutant type I TGF-b receptor that is incapable of recognizing Smad3 but still retains its kinase activity. With the aid of this key reagent, we were able to show that TGF-b can signal through a separate conduit via p38 MAPK to control apoptotic gene response. This finding provided a biochemical proof for the long-suspected Smad-independent TGF-b receptor signaling.
Currently we are pursuing the missing link between the TGF-b receptor and activation of p38 MAPK in this alternate signaling route. I believe these studies will result in our being able to paint a comprehensive picture of how TGF-b signals to control its target genes, a necessary step in our quest for manipulating this pathway for therapeutic goals.
The diverse biological functions exerted by TGF-b demand that its signaling pathway have built-in mechanisms for the integration of multiple cell-signaling inputs. The Smad-ubiquitin-factor, or Smurf, is one such regulatory node.
Based on interaction and biochemical characterization data, we and others have found that mammalian genomes carry two Smurf genes capable of directing Smads and the type I TGF-b receptor to proteasome-mediated degradation.
To assess the biological significance of this regulation, we created mouse null alleles for both Smurf1 and Smurf2 by homologous recombination. The Smurf1-null mice are viable but display an age-dependent increase in bone mass due to elevated osteoblast activity.
Although this phenotype is consistent with the activation of the bone morphogenic protein pathway, neither the level of Smad1 nor the level of its receptor was changed in the Smurf1-null mice. Instead, Smurf1 removal resulted in MEKK2 activation, which in turn caused the increase in osteoblast function.
It is possible that the MEKK2 loop is part of the Smad-independent signaling mechanism and that the deficiency of Smurf1 in Smad degradation was compensated by Smurf2. We are currently testing these hypotheses by characterizing mice deficient in Smurf2 alone or in combination with Smurf1.
In the third line of inquiries, we created several lines of mouse liver-tumor models expressing different Smad3 transgenes. We showed that forced expression of Smad3 protects the liver from chemically induced carcinogenesis, and we demonstrated that this protection was afforded by the ability of Smad3 to execute the TGF-b–induced apoptotic instruction by downregulating the expression of Bcl-2.
This
finding sheds light on the molecular mechanism that controls the liver-tumor
growth and offers new insight in our pursuit of novel approaches to treat liver
cancer. ![]()