
Mark
Connors
| T H E N I H C A T A L Y S T | M A Y – J U N E 2005 |
|
|
|
| P E O P L E |
RECENTLY TENURED
|
|
Mark
Connors
|
Mark Connors received his M.D. from Temple University in Philadelphia in 1985 and completed his pediatrics residency and chief residency at Tufts-New England Medical Center in Boston. He then joined NIAID in 1989 as a fellow in the Laboratory of Infectious Diseases. In 1993, he began a year of infectious diseases training at the Clinical Center and Childrens Hospital of Philadelphia. He returned to NIAID to the Laboratory of Immunoregulation in 1994 and is currently a senior investigator in the Clinical and Molecular Retrovirology Section.
Despite prolonged investigation, the fundamental mechanisms by which the immune response might control HIV are poorly understood. This area remains one of the most important in HIV research. A better understanding of the breadth and magnitude of effective HIV-specific immune responses, the HIV protein targets of these responses, and the mechanisms by which protection or control occur may provide insights critical for the development of effective immunotherapies and prophylactic or therapeutic vaccines.
To provide a better understanding of the basis of immunologic control, we have recruited a cohort of very rare patients that represent the best example available of a successful immune response to HIV and studied these patients’ responses in extreme detail.
The patients, referred to as long-term nonprogressors (LTNP), maintain immunologic restriction of HIV replication despite prolonged infection. It is likely their study holds important clues to the components of an effective immune response to HIV. Conversely, they also hold important clues regarding how control of HIV replication is lost in most infected individuals.
Prior definitions of LTNP have varied, were based on peripheral blood CD4+ T cell counts alone, and tended to delineate highly heterogeneous cohorts. Use of HIV replication circumscribes a much smaller, more homogeneous group of patients. This subgroup of LTNP patients—without the use of antiretroviral therapy—manages to maintain normal CD4+ T cell counts and plasma viral RNA below the level of detection of current widely used assays (<50 copies/mL plasma). Many of these patients have been infected for up to 20 years with no CD4+ T cell decline. It is 17 of these patients that we have now assembled as a unique cohort with nonprogressive disease. Our studies compare cells from these patients with those from MHC-matched and -mismatched control patients whose disease is progressing. Through these comparisons, we are systematically dissecting the mechanisms of immune-mediated restriction of HIV replication.
Many mechanisms have been proposed to explain the inability of cell-mediated immunity to control HIV replication in the majority of infected patients. These include viral factors and quantitative and qualitative factors within the HIV-specific CD8+ T cell pool. We have examined many of these parameters and our work has provided some surprising and counterintuitive answers.
For example, we have found that approximately 95 percent of the LTNP in our cohort carry the same MHC allele–HLA B*5701. The HIV-specific CD8+ T cell responses of these individuals is completely focused on peptides presented by the B5701 molecule, providing a functional link to this genetic association. But the HLA B*5701 allele alone is not sufficient to confer restriction of HIV replication. Progressors with this allele occur at expected frequency and have very high viral loads.
We also rule out the possibility that immunologic control in LTNP is based on the epitopes targeted, viral mutations, or higher numbers of HIV-specific cells. Most patients, LTNP and progressors, maintain very high percentages of CD8+ T cells in peripheral blood that are HIV-specific (10–40 percent). The persistence of such high frequencies of HIV-specific T cells—with or without immunologic control—indicates that the ability of LTNP to control HIV is based on qualitative rather than quantitative aspects of the immune response. These data also suggest that we are missing some fundamental parameters that govern this control.
Thus far we have found one qualitative parameter that distinguishes HIV-specific CD8+ T cells of LTNP from those of progressors. This is their ability to proliferate in response to HIV-infected cells in vitro. Whereas the CD8+ T cells of progressors divide poorly in response to HIV-infected cells, those of LTNP expand rapidly and synchronously. This expansion is paralleled by, or is coupled to, production of perforin, a molecule critical for cell-mediated killing of infected cells.
A deeper understanding of the basis of immunologic control in LTNP and the loss of immunologic control in progressors is likely to provide information that is critical to harness cellular immune response for immunotherapies or prophylactic vaccination. It will be very important to understand how qualitative changes in the HIV-specific response occur, whether they can be avoided in vaccinees that become infected, and whether they are reversible. In addition, we are looking for genes in LTNP that may have modified the HIV-specific response to permit such potent immunologic control over such a long period of time.
 |
|
Traci
Hall
|
Traci Hall received her Ph.D. in 1992 from the Department of Pharmacology and Molecular Sciences at the Johns Hopkins School of Medicine in Baltimore. After studies with Mette Strand on the parasitic disease schistosomiasis, she was an American Association for the Advancement of Science diplomacy fellow at the U.S. Agency for International Development. In 1994, she began a postdoctoral fellowship with Daniel Leahy in the Department of Biophysics and Biophysical Chemistry at the Johns Hopkins School of Medicine. In 1998, she joined NIH as a tenure-track investigator at NIEHS and is currently leader of the Macromolecular Structure Group in the Laboratory of Structural Biology.
My laboratory studies the mechanisms of post-transcriptional gene regulation, using structural and biochemical techniques to understand how gene expression is controlled after a messenger RNA (mRNA) is produced.
I was drawn to this area of research because of the importance of post-transcriptional gene regulation during embryonic development. In addition, post-transcriptional gene regulation is important for normal cellular processes. For example, regulation by microRNAs—which are small noncoding RNAs—falls into this category of gene regulation, and recent predictions suggest that about 30 percent of vertebrate genes may be regulated by microRNAs. My lab is particularly interested in understanding how protein-RNA interactions affect the function of proteins that control gene expression.
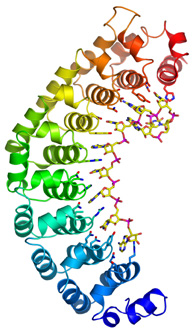 |
|
Ribbon
drawing of human Pumilio1 protein bound to Nanos Response Element RNA
(stick model).
|
Two examples from our work illustrate the diverse ways in which proteins can recognize RNA. In collaboration with Phillip Zamore’s laboratory at the University of Massachusetts Medical School in Worcester, we examined the structure and RNA-binding characteristics of a Pumilio protein. Pumilio proteins are important for stem-cell maintenance and differentiation in many organisms. They work by binding specifically to sequences in target mRNAs, downregulating expression of the protein.
We determined the crystal structure of the human Pumilio1 protein, both alone and bound to a high-affinity RNA ligand. Pumilio proteins comprise eight sequence repeats. Our structures showed that these repeats bind to the RNA, one base per repeat. We were amazed when we examined the details of the protein-RNA interaction and found that there appeared to be a "code" for sequence-specific recognition. Three protein side chains interacted with each RNA base, and particular sets of protein side chains seemed to recognize specific bases. For example, glutamine, asparagine, and tyrosine recognized uracil, or glutamine, cysteine, and arginine recognized adenine.
This result suggested that we could make point mutations in the protein and predictably alter the RNA-binding specificity. We tried this and it worked! We created a mutant protein that preferred the new RNA sequence over the original sequence. Our work suggests that Pumilio may be the first RNA-binding protein to use a true code to read RNA sequence. We are continuing to design additional mutant proteins to target particular RNA sequences.
In a second example, we examined the structure and binding specificity of a plant viral protein known as p19 that suppresses RNA silencing by binding to small interfering RNAs (siRNAs). This was in collaboration with József Burgyán’s laboratory at the Agricultural Biotechnology Center in Gödollö, Hungary. Our crystal structure of p19 protein in complex with a 21-nucleotide (nt) siRNA revealed that the protein forms a dimer to interact with the RNA and uses two sets of tryptophan side chains to clamp the ends of the double-stranded middle of the siRNA. It suggested immediately that p19 recognizes the siRNA by measuring the length of the duplex RNA.
We tested this by measuring the binding of the protein to siRNAs of different lengths. We found that p19 binds poorly to siRNAs of only 19 nt, tightly to siRNAs of 20–22 nt, and progressively weaker to siRNAs as the length increases from 23–26 nt. Thus, p19 is specific for a particular size of RNA. In plants this is important because siRNAs come in two sizes, ~21 nt and ~24 nt. Thus, p19 inhibits processes directed by the smaller class such as mRNA degradation, but does not appear to affect processes directed by the longer class such as DNA methylation.
It has been fascinating to study Pumilio, which recognizes RNA based on sequence, and p19, which recognizes RNA based on size. We will continue to use structural and biochemical approaches to study RNA silencing and other mechanisms of post-transcriptional gene regulation and hope that future projects will yield results as fun and illuminating as these.
 |
|
Matthew
Kelley
|
Matthew Kelley received his Ph.D. from the University of Virginia, Charlottesville, in 1993. After a postdoctoral fellowship at the University of Washington, Seattle, he became an assistant professor at Georgetown University in Washington, D.C., in 1996. In 2000, he joined the Laboratory of Cellular Biology at NIDCD. He is now a senior investigator and head of the Section on Developmental Neuroscience.
One of the most intriguing events in developmental biology is the generation of cellular diversity. All complex organisms include groups of cells with distinct functions and phenotypes. In most cases, these specialized cells arise from undifferentiated progenitor cells that can assume different phenotypes in response to both intrinsic and extrinsic cues.
A better understanding of the factors that influence cells to develop as one cell type versus another will lead to a better understanding of the bases for different congenital disorders and provide valuable insights into the signaling pathways that must be activated for therapies based on stem cells.
In my laboratory, we investigate the development of different cell types within the highly invariant cellular mosaic of the sensory epithelium of the mammalian inner ear. Several factors led to the selection of this structure for my studies. First, the inner ear sensory epithelium comprises six different cell types that are generated in specific ratios to one another and are then arranged into a specific cellular pattern.
Because the number of cells and their pattern are so regular, defects in the pattern, even very subtle ones, are easily identified. In addition, progressive loss of cells within the sensory epithelium leads to deficits in hearing. Although interventions such as hearing aids and cochlear implants are available, it would obviously be preferable to restore normal function.
The cells within the sensory epithelium of the inner ear can be grossly divided into mechanosensory hair cells, which transduce sound waves into nervous impulses, and nonsensory supporting cells, which surround the hair cells and fill crucial anatomical and physiological roles within the epithelium.
Research from my laboratory demonstrated that virtually all of the cells within the developing epithelium initially attempt to develop as hair cells through the activation of a specific transcription factor called Math1. However, as development continues, individual cells compete with one another to maintain expression of Math1. Ultimately, some cells win this competition, increase expression of Math1, and go on to develop as hair cells. Subsequent experiments in the laboratory demonstrated that the number and position of cells that ultimately maintain expression of Math1, and therefore develop as hair cells, is dictated through the downregulation of a group of genes, called Ids, which function to dampen the activity of Math1.
As developing hair cells increase their expression of Math1, they generate extrinsic signals that act to shut down expression of Math1 in neighboring cells. At the same time, hair cells generate a second signal that induces these same neighbors to develop as supporting cells. We have not identified this second signal yet, but hope to in the near future.
A second research emphasis in the laboratory is determining the molecular factors that lead to the uniform orientation of cell types within the ear. All hair cells are only sensitive to vibrations in a single plane, so the correct orientation of each cell is crucial for normal auditory function. There are other examples of uniform orientation among groups of cells, but the molecular pathways involved have not been determined.
We recently identified the first genes that are required for uniform orientation in vertebrates, and we are now using these genes as probes to further understand the signaling pathways that coordinate these events.
Our results are beginning to point to signaling pathways and cellular interactions that are required to generate the precise cellular pattern within the mammalian inner ear. We hope to generate a more complete model of how this structure, and other complex structures, are assembled from pools of undifferentiated progenitor cells.
 |
|
Sue
Priola
|
Sue Priola received her Ph.D. in microbiology and immunology in 1990 from the University of California, Los Angeles, for studies on the molecular mechanisms of herpesvirus latency. In 1991, she joined NIAID at the Rocky Mountain Laboratories (RML) campus, where she completed her postdoctoral work on the transmissible spongiform encephalopathies. She was converted to a tenure track position in 1998 and is currently a senior investigator at RML.
I study transmissible spongiform encephalopathy (TSE, or prion) diseases. These are infectious, rare, and fatal neurodegenerative diseases of mammals. One of the most intriguing aspects of TSE diseases is that they have no known viral or bacterial component. Rather, conversion of the normally protease-sensitive mammalian prion protein (PrP-sen) into an abnormal protease-resistant form (PrP-res) appears to be at the center of events that occur during TSE infection, and PrP-res has been proposed as the TSE infectious agent. The fact that a cattle TSE—bovine spongiform encephalopathy (BSE)—has infected humans and concerns that chronic wasting disease of deer and elk in the United States may do the same underscore the importance of understanding how these diseases work.
Susceptibility to TSE infection can be influenced by amino acid homology between PrP-sen and PrP-res. Because the amino acid sequence of PrP-sen differs between mammalian species, I reasoned that these differences could be the basis for species-specific barriers to TSE infection. My laboratory used cell-associated and cell-free assays to study the role of the PrP amino acid sequence in PrP-res formation for three different mammalian species. We found that a single amino acid mismatch between PrP-sen and PrP-res, as well as post-translational modifications to PrP-sen, influenced species-specific PrP-res formation, suggesting a molecular mechanism for TSE species barriers that involves PrP-sen/PrP-res homology.
There are no in vitro systems to monitor early events that occur during TSE infection, and very little is known about how they infect cells. To address this issue, my laboratory recently developed a novel tissue culture system that enables us to monitor PrP-res formation immediately after exposure of cells to TSE. Using this system, we found that TSE infection is at least a two-stage process: 1) an initial stage of PrP-res formation that is independent of cell type and TSE strain and 2) a second cell-type– and TSE strain–dependent stage leading to persistent PrP-res formation and infection. Our results provide a basis for why TSE infection in vivo is restricted to certain cell types and suggest that cell-specific factors are involved that may provide new targets for TSE treatment. Currently, we are working to identify these factors.
One surprising result of these experiments was the finding that NIH3T3 mouse fibroblasts, long assumed to be resistant to TSE infection, could be persistently infected with mouse TSE agents. These results have rekindled concerns in the vaccine industry that even non-neuronal vaccine-producing cell lines exposed to substrates contaminated with the BSE agent could potentially become infected.
Our new tissue culture system also allowed us to address an unresolved question in TSE biology: How can an infectious agent with no known nucleic acid component have biologically distinct strains? We found that certain strain characteristics are the result of a complex interaction between PrP-sen, PrP-res, and the host cell. Currently, our hypothesis is that the cellular microenvironment where PrP-res formation occurs influences TSE strain characteristics, and we are working to identify these cellular microenvironments. For me, the most exciting aspect of this work is that it may help to explain some of the unique in vivo pathological phenotypes associated with different TSE strains.
In addition to understanding the molecular basis of TSE pathogenesis, my laboratory is also actively pursuing effective TSE therapeutics. We found that cyclic tetrapyrroles, a vast group of compounds that had never been tested in vivo against any infectious disease, are potent TSE inhibitors when given prophylactically (Science 287:1503–1506, 2000). These results are particularly exciting given the possibility that, because of
 |
|
Barbara
Rehermann
|
Barbara Rehermann earned her M.D. from Medizinische Hochschule in Hannover, Germany, in 1991. Her research interests started with a doctoral thesis in T cell receptor signaling at the same university from 1987 to 1988 and a research year on lymphocyte biology at Memorial Sloan-Kettering Cancer Center in New York from 1988 to 1989. After internship and residency at the Universities of Essen and Medizinische Hochschule from 1991 to 1993, she completed a postdoctoral fellowship in viral immunology at The Scripps Research Institute, La Jolla, Calif., from 1993 to 1995. She then returned to Hannover, set up her own research laboratory, and received the Venia Legendi (an academic title in the German system) for Immunology at the same university. In 1998, she joined the NIDDK intramural program as a tenure-track investigator and is now a senior investigator in the Liver Diseases Branch, NIDDK.
My research program focuses on the immunology of viral and autoimmune liver diseases. A major interest is the immunology of hepatitis C virus (HCV) infection. Over the last 10 years, my laboratory has studied components of successful immune responses, as well as mechanisms of immune evasion in HCV infection.
We are using multiple approaches. First, we study immune responses of clinically well-characterized patient cohorts who are prospectively followed in the Liver Diseases Branch.
Second, we use the chimpanzee model, the only animal susceptible to HCV infection, to study virus-host interaction in the early phase of infection and at the site of viral replication—the liver. In this model, we use clonal HCV (HCV-RNA transcribed from HCV cDNA clones) and a unique set of immunologic reagents that we have generated based on the sequence of the infecting HCV and the chimpanzees’ MHC haplotype. The model provides the opportunity to rechallenge recovered animals and to test experimental immunotherapies in persistently infected animals.
Finally, we use transgenic mouse models to study basic immunological mechanisms, such as liver-specific auto-immunity, proteasomal processing of viral antigens, and the role of cross-priming in the induction of specific T cells.
Studying a single source outbreak of HCV, we were the first to demonstrate that HCV antibodies may disappear in approximately 40 percent of patients 10 to 20 years after recovery, whereas HCV-specific CD4+ and CD8+ T cells remain readily detectable in the circulation.
In collaboration with Jake Liang, Jay Hoofnagle, and Theo Heller in the Liver Diseases Branch, we subsequently demonstrated HCV-specific T cell responses in the blood of health-care workers shortly after accidental needlestick exposure to HCV-contaminated blood, even when HCV-specific antibodies remained undetectable. HCV-specific T cell responses are also often found in family members of HCV-infected patients and injection drug users who are frequently exposed to the virus but do not show any other evidence of past or present infection.
Collectively, these results suggest that the incidence of recovery from hepatitis C may be underestimated because HCV-specific antibodies may be lost after recovery or never induced, and cellular immune responses are rarely studied.
In collaboration with virologists Stephen Feinstone at the FDA and Charles Rice at Rockefeller University in New York, we then asked the question whether these HCV-specific memory T cells can be protective.
When we rechallenged spontaneously HCV-recovered chimpanzees with homologous and in one case heterologous HCV, we observed an attenuated course of infection with significantly reduced viremia and rapid HCV clearance without elevation of liver enzymes. Intrahepatic and peripheral HCV-specific memory T cell responses, but not antibody responses against HCV envelope proteins, correlated with HCV clearance and needed to be maintained to prevent HCV from becoming active again.
Thus, HCV-specific T cells, although unable to completely destroy the virus, appear to contribute to rapid control and clearance of the challenge inoculum.
These results have prompted us to evaluate strategies to induce novel T cell responses or to modulate and enhance preexisting but apparently ineffective HCV-specific T cell responses in patients who are persistently infected and seem unable to clear the virus spontaneously.
We have, for example, recently used a self-replicating form of recombinant cytopathic pestivirus RNA to express HCV antigens in murine dendritic cells (DCs). In this model, induction of cell death by the cytopathic replication resulted in antigen transfer from vaccine DCs to endogenous DCs in lymphoid organs of vaccinated mice, direct and cross-priming of T cell responses, and protective immunity in a surrogate virus challenge.
These studies are closely
related to our current efforts to decipher mechanisms of HCV persistence, such
as escape by mutation, adaptation to protea-somal antigen processing, and alteration
of dendritic cell and B cell functions. We expect that understanding these mechanisms
will enable us to induce protective immune responses via immunotherapy.![]()