
Jenny
Hinshaw
| T H E N I H C A T A L Y S T | J U L Y – A U G U S T 2003 |
|
|
|
| P E O P L E |
RECENTLY TENURED
|
|
Jenny
Hinshaw
|
Jenny Hinshaw received her Ph.D. from Brown University, Providence, R.I., in 1990 and did her postdoctoral work at The Scripps Research Institute in La Jolla, Calif. In 1995, she became a tenure-track investigator at NIH in the Laboratory of Cell Biochemistry and Biology, NIDDK, where she is currently a senior investigator.
My laboratory is interested in the dynamic process of membrane trafficking within eukaryotic cells. This process involves numerous specialized protein complexes and lipid domains. Over the past several years we have focused on one particularly intriguing set of proteins, the dynamin family of mechanochemical enzymes—a family of large GTPases that are potentially involved in nearly all cellular membrane stabilization and fission events. Dynamin itself is essential for receptor-mediated endocytosis, caveolae internalization, and trafficking to and from the Golgi.
 |
| Zhang, P. & Hinshaw, J.E.. Nat. Cell Biol. 3:922-926 (2001) |
|
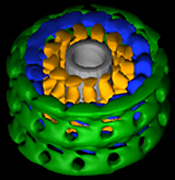
Three-dimensional reconstruction of dynamin in the constricted state. Three prominent densities—head, stalk, and leg—are shown in green, blue, and gold, respectively. The inner lipid leaflet is gray. |
I first became curious about the role of dynamin in endocytosis when, as a postdoctoral fellow in Sandy Schmid’s laboratory at Scripps, I discovered this protein was capable of self-assembling into ordered spirals. This work led to the hypothesis that dynamin assembles around the necks of coated pits in a spiral prior to membrane fission in endocytosis. The positioning of dynamin at the penultimate step in membrane fission further suggested that dynamin may play a direct role in formation of vesicles.
Soon after I arrived at NIH, my lab demonstrated that dynamin undergoes a GTP-dependent conformational change causing constriction and fragmentation. We believe this represents a critical step in the process that occurs when clathrin-coated pits bud from the plasma membrane. This ability of dynamin to constrict and generate a force on the underlying lipid bilayer makes it unique among GTPases as a mechanochemical enzyme.
To determine the conformational changes that occur during dynamin-induced constriction, we then calculated the first three-dimensional map of dynamin in the constricted state using cryoelectron microscopy and helical reconstruction methods at a resolution of 20 angstroms. The 3-D map consists of a repeating T structure (dimer) along the tube axis, which can be divided into three distinct density peaks.
Based on previous biochemical results and the docking of X-ray crystal structures into our 3-D map, we were able to predict the location of each of dynamin’s five distinct domains (GTPase, middle, Pleckstrin homology, GTP effector domain [GED], and proline-rich domain). Further analysis of the 3-D structure of dynamin allowed us to propose a mechanism of dynamin-induced membrane constriction: An interaction between GED and a GTPase domain from a neighboring dimer would lead to a decrease in both radial diameter and helical pitch.
In the near future, we hope to determine whether a common mechanism of action exists among all the dynamin family members. Additional dynamin family members have been implicated in numerous fundamental cellular processes, including other membrane fission events, antiviral activity, cell plate formation, and chloroplast biogenesis.
For example, a dynamin-related protein (Drp1) has been shown to localize to regions of mitochondria scission, and a plant dynamin family member, Arc5, has been elegantly shown to be involved in chloroplast division. Among all the dynamin proteins, self-assembly and oligomerization into ordered structures (such as rings and spirals) is a common characteristic and, for the majority, essential for their function. While they are continually being implicated in diverse functions of the cell, we would predict that all dynamin family members undergo similar conformational changes upon GTP addition.
Overall, understanding this unique family of large GTPases will provide valuable insights into the mechanism of fundamental cellular processes such as membrane fission and remodeling events.
 |
|
Brian
Kelsall
|
Brian Kelsall received his M.D. degree from Case Western Reserve University in Cleveland in 1986. He did an internal medicine residency at New York Hospital and an infectious disease fellowship at the University of Virginia in Charlottesville before joining the Laboratory of Clinical Investigation, NIAID, in 1992 as a postdoc in the Mucosal Immunity Section. He is currently a senior investigator in the Laboratory of Clinical Investigation, NIAID.
The mucosal immune system is marked by the necessity for precise regulation of positive and inhibitory immune responses. A host must respond to invading pathogens with the production of protective antibodies and cell-mediated immune responses, while, at the same time, controlling unnecessary responses to the myriad harmless foreign antigens that cross mucosal surfaces.
The major long-term goal of my laboratory is to develop a fundamental understanding of the factors that regulate the induction and maintenance of these disparate mucosal immune responses and to apply this knowledge to the development of novel mucosal vaccine strategies and treatments for mucosal inflammation.
After completing my internal medicine residency and a fellowship in infectious diseases that focused on mucosal immunity to Entamoeba histolytica, I came to NIH in 1992. As a postdoctoral fellow in Warren Strober’s lab, I identified several populations of dendritic cells (DCs) in Peyer’s patches, lymphoid structures that are primary sites for the induction of immune responses to antigens and microorganisms in the small intestine. DCs are now known to be a family of cells that are the major cells that present antigens to naïve T cells during a primary immune response. I demonstrated the importance of these cells for the induction of T-cell responses to orally administered antigens.
During my time as a tenure-track investigator, members of my laboratory demonstrated that DCs isolated from the Peyer’s patch are unique among DCs from nonmucosal lymphoid organs in their ability to produce IL-10 and induce the differentiation of IL-10–producing T cells. IL-10–producing T cells can potently inhibit immune responses, suggsting that mucosal DCs are conditioned to induce such regulatory T cells.
We went on to identify four different subpopulations of DCs in the Peyer’s patch, to study their phenotype and function, and to localize these populations to discrete regions of the Peyer’s patch. With this information, we developed models for how different DC populations are involved in tolerance to oral antigens ("oral tolerance") and the induction of immunity to pathogens.
Most recently we applied this knowledge to study the role of DCs in immunity to murine reovirus. We discovered that submucosal DCs are able to process reoviral antigens directly from overlying infected apoptotic epithelial cells. In addition, we identified novel receptors on human DCs that are responsible for the direct uptake of reovirus and possibly other viruses. We are currently exploring ways to exploit these findings for the development of mucosal vaccines that target DCs.
A second focus of the lab, which relates directly to our studies of mucosal immunity, is the regulation of cytokine production by DCs and macrophages. In particular, we demonstrated that signaling via several disparate surface receptors can inhibit the production of cytokines, such as IL-12, that are key in driving T-cell differentiation into Th1 cells. Th1 cells are important for host defense as well as for pathologic intestinal inflammation in certain types of inflammatory bowel disease, such as Crohn’s disease.
Translational leads are beginning to emerge from this work. For example, we showed that antibodies to and natural ligands of the b2-integrin CD11b/CD18 (complement receptor 3; CR3) can inhibit IL-12 production from human monocytes in vitro and suppress Il-12–dependent interferon-g production in a mouse model of septic shock. Then, most recently, we demonstrated that anti-CR3 can be used for the treatment of ongoing intestinal and skin inflammation in murine models.
We also demonstrated a primary role for G-protein–coupled receptor signaling in the regulation of cytokine production from both DCs and monocyte/macrophages. G-proteins are divided into subfamilies (such as Gs and Gi) based on their particular signaling characteristics. For signaling by the Gs subfamily of G-proteins, we demonstrated that cholera toxin, which directly activates Gs, can suppress IL-12 production from antigen-presenting cells. For signaling by the Gi subfamily of G-proteins, we observed that Gi-signaling by the chemoattractants C5a and fMLP has a similar suppressive effect.
We went on to show the importance of the Gi2a G-protein subunit for this effect. Gi2a-/- mice have exaggerated Th1 responses to most stimuli and develop spontaneous Th1-mediated intestinal inflammation. We are currently exploring the intracellular mechanisms by which G-proteins regulate IL-12 production and the in vivo mechanisms responsible for the exaggerated Th1 responses in the Gi2a-/- mice.
In summary, my primary research interest is understanding how immune responses are generated and maintained at mucosal sites like the intestinal tract. My hope is that information generated by our studies will result in the development of novel vaccine strategies for infectious diseases and novel ways to treat inflammatory bowel disease.
 |
|
Karl
Pfeifer
|
Karl Pfeifer received his Ph.D. in biology in 1988 from the Massachusetts Institute of Technology in Cambridge. His postdoctoral training was with Shirley Tilghman at Princeton University in Princeton, N.J. In 1995, he joined NICHD and there began the Section on Genomic Imprinting, where he is now a senior investigator.
Genomic imprinting represents a curious defiance of normal Mendelian genetics. Mammals inherit two complete sets of chromosomes, one from the mother and one from the father, and most autosomal genes will be expressed from both the maternal and paternal alleles. Imprinted genes, however, are expressed from only one chromosome, in a parent-of-origin-dependent manner.
Because silent and active genes are present in a single nucleus, the differences in transcriptional activity cannot be explained by the presence or absence of critical transcription factors. Instead, the transcription of imprinted genes represents a clear situation in which epigenetic mechanisms restrict gene expression. Imprinting, therefore, is a great model system for looking at the role of DNA modifications and chromatin structure in maintaining developmentally appropriate patterns of gene expression.
Imprinting is also of interest because imprinted genes are frequently associated with disease and developmental disorders. This is due to two unique vulnerabilities: First, imprinted genes are in some sense functionally haploid. For example, mutations in a maternal-specific gene, such as p57KIP2, will be completely dominant when on the maternal chromosome. However, the same mutation will be completely recessive when paternally inherited, thus allowing a way for the allele to persist in the population.
Second, imprinted genes are susceptible to disruptions in the imprinting process itself. If a cell loses track of the parental origins of its chromosomes, imprinted genes will be twofold overexpressed or completely unexpressed.
Our section is investigating a cluster of genes on the distal end of mouse chromosome 7. This cluster includes the H19 and Igf2 genes, which share developmentally complex patterns of gene expression but are reciprocally imprinted. H19 is expressed only from the maternal chromosome while Igf2 is paternal-specific.
We have focused on using genetic analyses with the goal of understanding three aspects of imprinting of these genes: establishment of the primary imprinting mark during gametogenesis, maintenance of this mark (or its secondary derivatives) during the many cell divisions and differentiation, and conversion of the imprinting mark into appropriate transcription patterns.
Our general approach has been to use molecular analyses to identify candidate DNA sequences that are likely to play a role in regulating gene expression and then testing the roles of these sequences by loss-of-function (knockout) and by gain-of-function (knockin) mutagenesis. Most importantly, we have defined a 2.4 kb region that lies between H19 and Igf2 that has three functions. First, it marks the chromosomal origin of the locus. Second, it functions as a DNA methylation-dependent transcriptional insulator that prevents expression of the maternal copy of Igf2. Third, it acts as a methylation-dependent transcriptional silencer that blocks expression of the paternal H19 allele.
Much of our current work is trying to understand the role of DNA methylation in imprinting. While such methylation clearly plays a role in regulating transcription of imprinted genes, we have been surprised to learn that its role in establishing the parental origins is much less certain. Identifying the true imprint, a mark that can be stably maintained during cell division but can be readily erased and reset each generation, is a key goal of our research.
We have also been interested in regulation and function of the Kcnq1 gene, also in the distal chromosome 7 cluster. This gene has great significance in medical genetics. Chromosomal translocations with breakpoints in the human KCNQ1 have been associated with Beckwith-Wiedemann syndrome (BWS), a fetal overgrowth syndrome, whereas point mutations in KCNQ1 are associated with long QT syndrome (LQTS), a predisposition to cardiac arrhythmias.
Our curiosity about this gene was initially raised when we noted that BWS but not LQTS is inherited in a parent-of-origin fashion. We have generated mouse mutant lines to understand the etiology of these two diseases. LQTS is associated with loss of function mutations in Kcnq1, with the phenotype only apparent postnatally. LQTS is not parent-of-origin dependent because Kcnq1’s imprinting is developmentally regulated: after birth, both alleles are expressed. The etiology of BWS appears to be quite a bit more complicated: It is not loss of KCNQ1 gene activity but a general disruption in normal embryonic imprinting patterns of several genes in the locus that is critical.
LQTS episodes in humans are very much induced by specific stresses, and we have also demonstrated this our mouse model. A major goal of our current Kcnq1 research is now directed toward understanding the molecular basis for this stress dependence.
 |
|
Allan
Saul
|
Allan Saul received his Ph.D. from the University of Queensland, Australia, in 1979, and then joined the Queensland Institute of Medical Research, Brisbane, pursuing the role of red cell polymorphisms on susceptibility to malaria and the development of malaria vaccines. From 1984 to 1986, he was a visiting scientist in the Laboratory of Parasitic Diseases, NIAID, returning to the Queensland Institute of Medical Research to head a Malaria and Arbovirus Unit. Over the next 13 years, he studied the epidemiology of malaria in Southeast Asia, the development of sustainable malaria control programs, and the development of malaria vaccines, culminating in a successful Phase 2 trial in Papua New Guinea. This trial was the first to show evidence of efficacy with a recombinant protein vaccine directed against blood-stage malaria parasites. In 2000, he joined the Malaria Vaccine Development Unit (MVDU), NIAID, where he is the co-director.
Malaria kills a million children each year and makes hundreds of millions of people sick. The parasite has become resistant to standard antimalarial drugs, and unfortunately, as new drugs are introduced, resistance soon follows. The lack of health care in many endemic areas also makes it difficult to deliver treatment, especially when that requires frequent medication. Malaria vaccines directed against more new targets could make a big difference in the battle against this costly disease.
We are developing two types of recombinant protein–based vaccines: one directed against blood-stage parasites that cause the disease, and the second against mosquito-stage parasites that transmit malaria from one person to another.
Several promising vaccine candidates have been identified; however, a major bottleneck has been getting these from the laboratory through Phase 1 safety and immunogenicity studies and Phase 2 studies in malaria-infected people to see whether they work.
Normally, a pharmaceutical company would accomplish these tasks, but, given the development effort required and the lack of a profitable market for malaria vaccines, public sector input is required. At the MVDU, we have developed something akin to a biotechnology company within the NIH to move the malaria vaccines through development. Our generic technology units provide the expertise for vaccine production and testing as well as product specialists who concentrate on individual vaccines.
We currently have six antigens either in clinical trials or undergoing the preclinical testing that may lead to trials. Several other antigens are in the pipeline and are likely to enter clinical trials in the foreseeable future.
We face two major technical challenges: formulation and human testing. In a vaccine, antigens are delivered in a formulation containing an adjuvant—a substance that stimulates the immune response. Although vaccines have been formulated with adjuvants for more than 70 years, the immunology underlying adjuvant activity is poorly understood. As a result, formulation is highly empirical. Developing formulations that will work with mixtures of antigens is even more complicated. We will put a major effort into studying adjuvant design and action to make this less hit-and-miss.
It takes a surprisingly large number of trials to test the vaccines we are producing. We plan about 30 Phase 1 trials in the United States over the next few years, leading to trials in countries where malaria is endemic. In collaboration with the University of Mali, we have developed a field-testing site in Mali, Bamako, and we will establish additional sites in other countries burdened with malaria .
Although the program is largely developmental, I am also pursuing several basic research questions. These include the nature of protective immunity in malaria; the impact of antigenic diversity on protective immunity; and the effect of new control measures, especially vaccines, on the population dynamics of malaria.
 |
|
Yun-Bo
Shi
|
Yun-Bo Shi received his Ph.D. from the University of California at Berkeley in 1998 under John E. Hearst and obtained his postdoctoral training under Donald D. Brown at the Carnegie Institution, Baltimore, MD. In 1992, he was recruited as a tenure-track investigator to head the Unit on Molecular Morphogenesis at NICHD. He is currently a senior investigator in the Laboratory of Gene Regulation and Development,
My laboratory has been interested in understanding the molecular mechanisms of tissue remodeling and organogenesis during p ostembryonic development in vertebrates. Despite enormous improvement made in our understanding of the molecular mechanisms governing embryogenesis from studies in various animal models, the progress in the study of postembryonic development has been hampered by the lack of proper developmental systems. We have chosen the thyroid hormone–dependent metamorphosis of Xenopus laevis as a model with which to study postembryonic development.
Amphibian metamorphosis is similar to postembryonic development in higher vertebrates in many aspects. An important advantage of this model system is its absolute requirement for thyroid hormone, which is also critical during the mammalian postembryonic period (from a few months before to a couple of years after birth in humans). Furthermore, this process takes place externally in free-swimming tadpoles. Thus, one can easily manipulate the process by simply changing thyroid hormone levels in the tadpole-rearing water or even in organ or primary cell cultures.
This model system allows one to avoid the complications associated with studies on fetal development in mammals, in which the fetuses enclosed in the uterus are inaccessible to manipulations. In the uterus, the effects of any treatments or manipulations may also trigger changes in both the embryos and the fetal environment due to maternal response, thus making it difficult to interpret the outcome.
Our research focuses on two early steps during Xenopus laevis metamorphosis by taking advantage of its total dependence on thyroid hormone. These are:
![]() How thyroid hormone receptors regulate thyroid hormone–response genes during
development
How thyroid hormone receptors regulate thyroid hormone–response genes during
development
![]() What roles the thyroid hormone–response genes play during tissue remodeling
What roles the thyroid hormone–response genes play during tissue remodeling
In the first area, we first carried out several in vitro and in vivo studies on the functions of thyroid hormone receptors and their expression profiles during development. These led us to propose a dual-function model for thyroid hormone receptors in development—that is, as transcriptional repressors when thyroid hormone is absent to ensure a proper growth period before metamorphosis, and as transcriptional activators when thyroid hormone is present to initiate the gene regulatory cascade that leads to metamorphosis.
To validate the model in vivo, we used the chromatin immunoprecipitation (ChIP) assay to study the receptor binding to endogenous target promoters in developing animals. We were able to show, for the first time in any developing animal, that thyroid hormone receptors bind constitutively to target genes in vivo.
Furthermore, using the same ChIP assay, we have shown that the binding of the receptor leads to local recruitment of co-repressor complexes and histone deacetylation in the absence of the hormone. Upon thyroid hormone treatment of the tadpoles, which induces metamorphosis, the co-repressors are released and co-activators are recruited, leading to local histone acetylation and gene activation. These studies provide direct in vivo evidence in a vertebrate developmental system for an important role of cofactor complex in chromatin remodeling and gene regulation by thyroid hormone receptors.
In the second area, we have identified and characterized a large number of genes activated by thyroid hormone during metamorphosis. In particular, we have provided strong support for the involvement in tissue remodeling of a group of thyroid hormone–induced genes encoding matrix metalloprotein-ases in cell fate determination (cell death vs. cell proliferation and differentiation). More importantly, we have shown in organ cultures that the matrix metalloproteinase stromelysin-3 is required for larval epithelial apoptosis and adult epithelial migration during intestinal remodeling.
Both thyroid hormone receptors and matrix metalloproteinases play important roles in human development and pathogenesis. However, it has been difficult to study the in vivo functions and the underlying mechanisms of these important proteins during postembryonic development due to the lack of good model systems. Thus, our findings are not only critically valuable for the understanding of frog development but also have important implications in higher vertebrates, including humans.
Our current research in both areas is directed toward functional analyses of these genes by using a relatively new transgenic approach in frogs to determine the roles of these genes in the transformations of different organs. To understand how these genes exert their effect on development, we plan to:
![]() Investigate the roles of different cofactors in gene regulation by thyroid hormone
receptors through a combination of molecular analysis in vivo and transgenic
studies
Investigate the roles of different cofactors in gene regulation by thyroid hormone
receptors through a combination of molecular analysis in vivo and transgenic
studies
![]() Isolate and functionally characterize substrates of matrix metalloproteinases
during metamorphosis.
Isolate and functionally characterize substrates of matrix metalloproteinases
during metamorphosis.
 |
|
James
Troendle
|
James Troendle received his Ph.D. from University of Maryland, College Park, in 1992 and joined the Biometry and Mathematical Statistics Branch of the Division of Epidemiology, Statistics, and Prevention Research, NICHD, as a staff fellow. He was converted to tenure track in 1997 and is now a senior investigator.
My primary research at NIH has been in developing methods for multiple hypothesis testing, while controlling an appropriate error rate. Simple methods based on the Bonferroni inequality allow one to safely test several hypotheses with control of the overall chance of a spurious finding. However, such methods lack power, largely because they fail to exploit the correlation between the test statistics. Furthermore, controlling the chance of a spurious finding is too stringent a requirement in some cases. In these cases, methods that allow for a few spurious findings can be much more powerful.
The effect of correlation on the conservatism of Bonferroni methods is potentially great. Such methods control the chance of a spurious finding regardless of the correlation. As a consequence, the methods become more conservative as the correlation increases.
My motivating example was an analysis of 55 minor malformation types in babies born to diabetic and nondiabetic women. In this analysis we wanted to know which of the 55 malformation types are associated with having a diabetic mother. To determine this, one must test all 55 hypotheses that a particular malformation is not associated with having a diabetic mother. Because the presence of one malformation type is correlated with the presence of another malformation type, Bonferroni multiple testing methods are conservative. Less conservative (and therefore more powerful) methods can be obtained by comparing the observed test statistics to the multivariate distribution obtained by permuting the vectors of malformation indicators. Permutation methods implicitly use the correlation by working with the intact vectors of outcomes. Doing so permitted us to conclude that two of the 55 malformations in the infants were linked to having a diabetic mother.
Recently, interest in multiple testing of this sort with data on gene expression from microarrays has increased dramatically. Each microarray can have up to about 10,000 gene expressions. In microarray studies, one typically does not mind having a few spurious findings because the results will have to be confirmed in later studies. In this case, multivariate permutations can be used to conduct multiple hypothesis testing (while controlling the number of spurious findings) far more powerfully than by using Bonferroni methods. With my colleagues Ed Korn, Lisa McShane, and Richard Simon of NCI, I have developed a method to perform such tests on microarray data. Software for the procedure is available in BRB-ArrayTools of NCI.
My current research is in extending permutation methods to allow for adjustment of covariates, while still controlling an appropriate error rate. For example, in the malformation analysis above it is known that certain malformations are associated with weight and that weight is in turn affected by diabetes. Therefore, it is important to see whether the malformations that are linked to diabetes are so only because of their relationship with weight. Adjustment for such covariates is important, especially in observational studies that can be greatly affected by confounding.
 |
|
Charles
Vinson
|
Charles Vinson received his Ph.D. from the University of Virginia in Charlottesville in 1987 and did postdoctoral work at the Carnegie Institution of Washington on the grounds of the Johns Hopkins University in Baltimore, Maryland. He joined the Laboratory of Biochemistry in NCI in 1991. He became a senior investigator in 2002 and is now chief of the Gene Regulation Section in the Laboratory of Metabolism.
My interests are in the area of the structure and function of B-ZIP transcription factors. There are approximately 60 genes in the human genome that contain the B-ZIP protein motif. Each monomer of the B-ZIP dimer is a long bipartite a-helix. The COOH-terminal half either homodimerizes or heterodimerizes via a leucine zipper coiled coil motif to bind sequence-specific DNA. The NH-terminal half binds DNA in a sequence-specific manner.
My research in NCI started with a detailed analysis of the amino acids that regulate the stability and dimerization specificity of B-ZIP proteins. We did a double-mutant thermodynamic analysis and calculated coupling energies for charged amino acids in the g and e positions that lie across the leucine zipper interface. More recent work has examined the contribution of amino acids in the a position to dimerization specificity. These data have allowed us to predict dimerization partners of all human B-ZIP proteins, predictions that have been recently verified using protein microchip technology.
We have used our insight into the leucine zipper structure to design a dominant negative protein that acts in a dimerization-specific manner to inhibit the DNA binding of B-ZIP proteins. Such a reagent would inhibit the function of all structurally related B-ZIP proteins and overcome the redundant functions of many mammalian proteins. The leucine zipper itself could be a possible dominant negative protein. But such a reagent does not work because DNA binding stabilizes the B-ZIP dimer.
To overcome this limitation, we designed an acidic amphipathic protein sequence to replace the basic region. These A-ZIP protein constructs form heterodimers with B-ZIP proteins. The designed acidic amphi-pathic protein forms a coiled coil with the basic region of the B-ZIP motif, stabilizing the heterodimer up to 5.0 kcal/mol per dimer. The A-ZIP dominant negatives can totally inhibit the DNA binding of a B-ZIP protein in an equimolar competition.
We are now expressing these dominant negatives in transgenic mice. Our first transgenic mouse expressed a dominant negative that inhibits both the C/EBP and AP-1 family of transcription factors using a fat-specific promoter. These mice are born with no fat tissue, the only such mouse yet reported. The metabolic consequences of having no fat are profound. The mice overeat because they do not have leptin, a fat-derived hormone that acts as a satiety signal, and this results in profound type 2 diabetes.
To have more experimental control over expression of these dominant negatives, we are presently using the tetracycline system that allows us to control expression of the dominant negative. We have generated mice that express A-FOS, A-CREB, A-C/EBP, and A-USF, dominant negatives that heterodimerize with and abolish the DNA binding of JUN, CREB, C/EBP, and USF respectively.
We can express these dominant negatives in a tissue-specific manner by crossing these mice to mice that express the tetracycline transactivator under the control of a tissue-specific promoter.
Expression of these dominant negatives in skin or heart during development results in lethality. Expression of these dominant negatives in the adult mice does not produce a phenotype. We are collaborating with the Stuart Yuspa group at NCI to study carcinogenesis in the skin of these mice.
We find that the A-CREB mice do not get tumors and the A-FOS and A-C/EBP mice produce sebaceous tumors instead of the typical papillomas. We are examining these models in more detail to understand how these dominant negatives are preventing malignant papillomas.
We have also expressed A-FOS in the striatum of the brain. In collaboration with Ron Paletzki, NIMH, we have demonstrated that these mice have enhanced sensitization and preference for cocaine. We are now modulating the expression of A-FOS during the sensitization paradigm to determine when A-FOS expression is critical for sensitization to cocaine.
We will continue to determine
whether expression of these dominant negatives ameliorates or exacerbates pathological
states in the adult. ![]()