
Fred
Dyda
| T H E N I H C A T A L Y S T | M A Y – J U N E 2002 |
|
|
|
| P E O P L E |
RECENTLY TENURED
|
|
Fred
Dyda
|
Fred Dyda received his Ph.D. from the University of Pittsburgh in 1992 in protein crystallography. He did his postdoctoral work at the Laboratory of Molecular Biology, NIDDK, where he is now a senior investigator.
I am interested in the function of complexes of macromolecules and why these complexes are always more than the sum of their parts. Our main experimental approach is to determine the three-dimensional structure of complexes using X-ray crystallography—the only experimental method that can resolve the structures of large complexes at near-atomic resolution. Unfortunately, this yields images—or structures—that are static, whereas macromolecules are inherently dynamic. To overcome this limitation, we attempt to determine several structures, representing different conformational forms or steps along a pathway. We then infer dynamics from the resulting array of static structures.
One problem we are studying is how the intracellular adaptor molecules, known as 14-3-3s, modulate conformational state and, through this, the biological activity of numerous binding partners. 14-3-3s bind their binding partners in a phosphorylation-dependent way.
One such binding partner is the melatonin rhythm enzyme, serotonin N–acetyltransferase. In collaboration with the laboratory of David Klein at NICHD, we have shown that this enzyme undergoes a major conformational rearrangement during catalysis involving a large movement of a surface loop. When the enzyme is phosphorylated, it forms a stable complex with 14-3-3. We were able to grow diffraction quality crystals of this complex and thus solve its three-dimensional structure. This is still the only structure ever obtained for a 14-3-3 bound to an active protein.
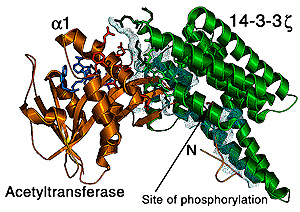 |
|
Detailed
interactions between serotonin N-Acetyltransferase
and 14-3-3. The dots (lighter areas) represent that part of 14-3-3’s
surface that interacts with the enzyme. A large portion of this surface
stabilizes helix a1 of the enzyme; several
turns of this helix forms from a loop during substrate binding.
|
Surprisingly, we found that 14-3-3 conformationally modulates exactly that loop of the enzyme that undergoes structural rearrangement during catalysis. Indeed, 14-3-3 stabilizes a conformational state of the enzyme with its substrate-binding site open, allowing easy access by both its substrates.
Our finding introduced a new way of thinking about the biological action of 14-3-3s, namely that rather than just simple binding to other molecules, their more significant action is changing the structure of their binding partners. It will be important to determine how far our results from serotonin N–acetyltransferase can be generalized. If conformational modulation is the rule in 14-3-3 biology, then how can 14-3-3 molecules act on a vast array of very different binding partners? To answer this question, we hope to analyze a series of structures of these 14-3-3 complexes with different enzymes and transcription factors.
We are also studying an elaborate DNA transposon called Tn7, work done in collaboration with the laboratory of Nancy Craig at the Johns Hopkins University (Baltimore)/Howard Hughes Medical Institute. This system is particularly intriguing because it requires the assembly of four different proteins for transposition. Tn7 is a clear example of the rule that a complex is much more than the sum of its parts, as each individual component is inactive for transposition on its own.
Still, even the isolated components are full of surprises. For instance, we have shown that one of the transposase molecules, TnsA, contrary to expectations, does not follow the paradigm that all transposases (such as HIV integrase, for example) share an RNaseH-like fold. Rather, TnsA looks like a type II restriction enzyme, making it a truly unique transposase. Based on what we have learned from TnsA, we are now in the process of assembling other complexes of Tn7 proteins in order to understand how the transposon moves antibiotic resistance genes between cells.
 |
|
Doug
Fields
|
Douglas Fields received his Ph.D. from the University of California, San Diego, in 1985 and did postdoctoral work at Stanford (Calif.) and Yale (New Haven, Conn) universities and NICHD. He is now a senior investigator in the Laboratory of Cellular and Synaptic Neurophysiology, NICHD, where he heads the section on Neurocytology and Physiology.
Driving Mr. Albert, by Michael Paterniti, tells the factual story of pathologist Thomas Harvey, who performed the autopsy on Albert Einstein in 1955 and then irreverently removed Einstein’s brain, took it home, and kept it hidden for nearly 40 years. Over the years, he doled out slices of it to scientists and pseudoscientists around the world who probed it for clues to Einstein’s genius. One respected scientist, Marian Diamond of the University of California at Berkeley, could find nothing unusual about the size or number of neurons in the slice of Einstein’s brain, but she noticed a large increase in the number of nonneuronal cells, termed glia. This observation is curious because traditionally glial cells are regarded as having only a passive support role in maintaining the extracellular environment. Research from my lab shows that glia can detect the electrical activity in neurons and respond physiologically and functionally to the signals.
My interest in neuron-glial communication developed from a more general interest in how development of the brain is modified by neural impulse activity in fetal and early postnatal life. I started my reasoning from the fact that information in the nervous system is coded in the pattern of neural impulse activity in the brain. Next I hypothesized that neuronal genes must be regulated by the pattern of impulse firing, because long-term changes in neuronal structure and function must involve the regulated expression of appropriate genes.
By stimulating neurons to fire in different temporal patterns and then measuring the amount of mRNA for genes known to be important in formation of neural circuits or in adaptation to the environment, we found that this was indeed the case. We observed that one could turn a particular gene on or off—for example, a cell adhesion molecule on the surface of axons that bundles them together into cables with other axons—simply by dialing up the correct stimulus frequency on our electrophysiological stimulator. The result was that the microscopic structure of the neurons changed accordingly: The axons either bundled or unbundled, depending upon the stimulus frequency we delivered to regulate the appropriate gene.
Having observed that neuronal genes could be regulated according to the pattern of neural impulse activity in developing neural circuits, we faced a deeper question: How could genes in the nucleus of the neuron be controlled by the pattern of electrical depolarization at the cell membrane? To study this, we used biochemical, molecular, and confocal calcium imaging to trace the stimulus transduction pathways from the cell membrane to the nucleus of the neuron in response to different patterns of impulse activity. What we observed was that temporal patterns of membrane depolarization are transduced and decoded within the cytoplasm and nucleus of neurons in accordance with differences in the kinetic response of individual signaling reactions comprising the network of intracellular signaling pathways. Some signaling pathways were sluggish and could not respond well to rapid bursts of impulses, but once they were activated, their lethargy in inactivating meant that they could sustain signals between bursts of action potentials that were separated by long intervals of inactivity. Thus, signals of different temporal patterns were propagated through distinct signaling pathways that were favorably tuned to those patterns of activation and ultimately regulated different transcription factors and different genes.
This work helps explain how the nervous system becomes modified by interaction with the environment, including some of the molecular mechanisms involved in encoding memory. Memory comes in two forms—short term and long term—and the transition from one to the other requires gene transcription and new protein synthesis. Your best friend’s name is stored in your long-term memory, for example, but the name of a person to whom you have just been introduced is stored in short-term memory and may be gone within a few minutes.
Prior to our work, it was theorized that an unknown signaling molecule must be generated when a synapse fires. The signaling molecule would wend its way from the synapse up the dendritic tree to the nucleus of the neuron to turn on the genes necessary for permanently strengthening the synaptic connection. Our work showed instead that when neurons in the hippocampus of the brain fire, signaling pathways originating from the cell membrane are activated, propagate to the nucleus, and regulate transcription factors and genes associated with permanent increases in synaptic strength and memory formation.
What does this have to do with Einstein’s glia? The intimate physical association between glial cells and neurons led me to wonder whether impulse activity in neurons might also be detected by glia and have some influence on their function. Using confocal calcium imaging to detect intracellular calcium fluxes in co-cultures of neurons and glia, our studies showed that when axons were stimulated to fire impulses, the glial cells nearby responded with large increases in cytoplasmic calcium concentration.
Through a series of pharmacological and biochemical experiments, we identified the signaling molecule mediating the communication between neurons and glial cells in the PNS (Schwann cells) and showed that neural impulse activity in axons could regulate glial genes, the mitotic rate of glia, and their differentiation. This would suggest that the distinctive architecture of Einstein’s brain resulted from the way his mind worked, rather than vice versa.
One of the key functions of Schwann cells is to form the myelin membrane that wraps around axons and insulates them to enable rapid conduction of nerve impulses. The importance of myelin can be seen in the growing abilities of a baby to hold its head up and eventually walk. It can also be seen in the deficits in adults when myelin is damaged through a disease such as multiple sclerosis. Our studies showed that impulse activity arrested the development of early Schwann cells and prevented the formation of myelin that might otherwise occur prematurely. This provided a way for myelination and development of Schwann cells to be coordinated according to functional activity in the nervous system.
In a series of pharmacological experiments, we identified the molecule that carried the signal from neurons to Schwann cells and concluded that it was ATP. Using the enzyme that allows fireflies to glow—a reaction that requires ATP—we were able to detect the release of ATP into the cell-culture medium when we stimulated axons to fire action potentials.
Sometimes a small amount of light can illuminate a new landscape.
 |
|
Stephen
Ikeda
|
Stephen Ikeda received his M.D. and Ph.D. from the University of Maryland School of Medicine, Baltimore, in 1980 and 1983, respectively. He did postdoctoral work at the NIAAA before joining the Department of Pharmacology and Toxicology at the Medical College of Georgia in Augusta and then joining the Guthrie Research Institute in Sayre, Pa., as a senior scientist and director of the Laboratory of Molecular Physiology. He also served as director of the Guthrie cDNA Resource Center and was an adjunct research professor of pharmacology, State University of New York Upstate Medical Center, Syracuse. He returned to NIAAA in February 2002 as chief of the Laboratory of Molecular Physiology.
The main focus of my laboratory is to understand signal transduction pathways underlying ion-channel modulation in neurons. In recent years, we have concentrated our efforts on understanding the molecular mechanisms involved with neurotransmitter-mediated modulation of N-type (Cav2.2) Ca2+ channels in sympathetic neurons. Because Ca2+ flux through the N- and P/Q-type (Cav2.1) Ca2+ channels triggers exocytotic release from presynaptic nerve terminals, modulation of neuronal Ca2+ channels is an important mechanism for fine-tuning synaptic transmission at both peripheral and central synapses. A few years ago, we discovered that a well-described mode of N-type Ca2+ channel modulation, termed voltage-dependent inhibition, was mediated by G protein bg subunits. Work from our lab and others defined a compact signaling pathway comprised of G protein coupling receptor (GPCR), G protein heterotrimer (and associated proteins), and the a1 subunit of the Ca2+ channel. Within this framework, we pursue several avenues of research, using electrophysiological, molecular, biological, and biochemical techniques.
Specificity of heterotrimeric G protein signaling components. Heterotrimeric G proteins are comprised of three subunits (Ga, Gb, Gg). Because there are numerous isoforms of each subunit, we wondered whether N-type Ca2+ channel modulation requires discrete Gabg combinations to serve specific functions. We are taking two separate, complementary approaches to address this question. The first is to reconstruct the signaling pathway using defined G protein subunits to determine which combinations are capable of supporting modulation. The second is to selectively ablate elements of the signaling pathway to determine the effect on pathways natively expressed in sympathetic neurons.
Role of RGS proteins in ion channel modulation. RGS (for "regulator of G protein signaling") proteins are a large, diverse family of proteins that interact with activated Ga subunits. The main result of this interaction is an acceleration of the GTPase activity of the Ga subunit. However, it is becoming clear that RGS proteins serve different specific roles in regulating the duration and strength of G protein signaling. We are currently investigating how RGS proteins affect modulation of ion channels.
(GPCR) coupling and pharmacology. The sympathetic neuron turns out to be an ideal surrogate for studying GPCRs normally expressed in central neurons. Heterologous expression of metabotropic glutamate receptors (mGluRs) in sympathetic neurons allows researchers to study molecularly defined mGluR isoforms and also mutated and epitope-tagged mGluRs within a neuronal environment. We are also interested in another GPCR—the CB1 cannabinoid receptor. We have previously shown that expression of CB1 in SCG neurons results in functional coupling to Ca2+ channels and that SR141716A acts as an inverse agonist in this system. Our current interest is in determining whether CB1 coupling to specific Ga subunits alters the pharmacology of the receptor. Specifically, we seek to determine whether endocannabinoid efficacy is influenced by the heterotrimeric G protein composition.
We are currently working on new methodologies, such as circularly permutated EYFP-based optical sensors to detect Gbg interactions and FRET-based approaches to Ca2+ channel/Gbg binding. We hope to extend some of our findings on GPC Rs and ion-channel modulation to more complex systems, such as synaptically coupled neurons.
 |
|
Steven
Sollott
|
Steven Sollott received his M.D. from the University of Rochester (N.Y.) School of Medicine and Dentistry in 1984. He completed residency training in internal medicine at Cornell in 1984 and a cardiology fellowship at Johns Hopkins University in 1991. He is board certified in internal medicine and cardiovascular disease. He worked as a fellow in the NIH Medical Staff Fellowship program and as an NIH senior staff fellow before being appointed to NIA’s tenure-track program. He also holds an appointment as an assistant professor in the Department of Medicine, Division of Cardiology, at Johns Hopkins. He is now a senior investigator in the Laboratory of Cardiovascular Science (LCS), NIA.
We are studying structure and function of cells from the cardiovascular system along three principal lines to gain an understanding of basic cell biological processes that may have implications for the pathophysiology and treatment of human disease. These lines are:
![]() Mechanisms of cardiac contractility
Mechanisms of cardiac contractility
![]() Nature and control of mitochondrial instability during oxidant stress
Nature and control of mitochondrial instability during oxidant stress
![]() Cellular changes after vascular injury
Cellular changes after vascular injury
How the heart adapts to stress is critical to the quality of life during periods of good health and to the ability to survive during periods of disease. Muscle stretch is a principal determinant of cardiac performance. Cardiac muscle stretch modulates contraction via enhancement of the excitation–Ca2+-release process, but how this occurs was largely unknown prior to our latest work. We found that myocyte stretch modulates the elementary Ca2+-release process from ryanodine-receptor Ca2+-release channels ("Ca2+-sparks") and the electrically stimulated Ca2+-transient.
Stretch induces PI3-kinase–dependent phosphorylation of both Akt and eNOS, resulting in localized NO production and a proportionate increase in Ca2+-spark frequency. We propose that myocyte NO produced by activation of the PI3-kinase–Akt-eNOS axis acts as a second messenger of stretch by enhancing Ca2+ release, contributing to myocardial contractile activation. The significance is that this mechanism could serve as a physiologic sensor of cardiac stretch by generating NO, providing a novel link between cardiac muscle length and excitation-contraction coupling.
Oxidant stress is an important factor in normal aging as well as in diseases such as atherosclerosis, heart attack, and stroke. Reactive oxygen species (ROS) are major contributors to oxidant stress. Mitochondria are both a major source of ROS and a target for their damaging effects.
A central event in apoptosis is a phenomenon known as the mitochondrial permeability transition (MPT). Various oxidants stimulate apoptosis, whereas antioxidants inhibit it, suggesting a role for ROS as initiators or downstream mediators of apoptosis. Because mitochondria are themselves the major intracellular sources of ROS production, together with the fact that ROS exposure and altered redox state can lead to the MPT, we hypothesized that under certain circumstances this biological system could become self-amplifying and unstable.
We developed a new model using "interactive" confocal microscopy, which enables controlled, incremental ROS photoproduction and accumulation in individual mitochondria inside living cardiac myocytes. Accumulation of ROS levels past a threshold reproducibly triggers abrupt mitochondrial depolarization, which we proved is the result of induction of the MPT. This model enables real-time observation of the initiation and induction of the MPT and its consequences in individual mitochondria inside living cells.
Thus, we were able to observe that this ROS-triggered induction of the MPT coincided with an immediate large burst of additional ROS generated by that same mitochondrion, together with a brief period of unstable Ca2+ handling in its immediate vicinity.
This phenomenon could also contribute to pathological disturbances in cardiac excitation and rhythm, for example, during postischemic reperfusion arrythmias, a major cause of sudden death after heart attack. For this newly described phenomenon accompanying induction of the MPT we coined the name mitochondrial ROS-induced ROS release (RIRR). We think that this link between MPT and RIRR could be a fundamental phenomenon in mitochondrial and cell biology and may be related to programmed mitochondrial destruction in cardiac myocytes, as well as programmed cell death (apoptosis).
One of the most common causes of serious heart malfunction—heart attack—is the result of underlying diseases of blood vessels, such as atherosclerosis. Thus, improvements in the treatment of coronary artery disease have had a significant positive effect on overall cardiovascular health. Intraarterial stents have become a primary therapy for treating coronary artery disease. Stents limit the elastic recoil and late vascular wall remodeling after angioplasty. When restenosis occurs in this scenario, it is an iatrogenic complication of the additional arterial injury caused by stent placement itself, causing abnormal migration and proliferation of vascular smooth muscle cells into the artery lumen, which obstructs normal blood flow.
Restenosis rates for patients undergoing stent placement procedures (more than 400,000 annually in the United States) result in clinical failure in about 20 to 30 percent of patients by 6 to 9 months. Thus, the anticipated clinical effect of drug-coated stents that would prevent cellular responses leading to restenosis is very significant.
Perhaps my most noteworthy discovery relates to the finding that paclitaxel (Taxol), a drug used to treat cancer, could markedly attenuate vascular restenosis after angioplasty. Together with colleagues at LCS and Johns Hopkins, we have organized follow-up studies based on the idea that paclitaxel could be of therapeutic value in preventing human restenosis with minimal toxicity. These studies were included in "Selected NIH Intramural Research Accomplishments 1993–2001" (The NIH Catalyst, May–June 2001).
Paclitaxel is now one of the two most promising treatments currently being tested in humans to prevent vascular restenosis after angioplasty. Human clinical trials are currently in progress in Europe, Asia, and the United States. The initial findings from two of the international paclitaxel-coated stent antirestenosis trials, ELUTES and TAXUS I, indicate that paclitaxel may prevent human restenosis without adverse effects.
Looking ahead, I hope to pursue studies that advance the understanding of how to prevent or limit pathological processes damaging to the heart. On the prevention front, we plan to continue to investigate the safety, feasibility, and efficacy of paclitaxel-coated coronary stents for the prevention of restenosis after coronary angioplasty. We will also pursue how to make the heart more resistant to ischemic damage by focusing specifically on mitochondrial protection, because these organelles are particularly susceptible to damage from oxidant stress, with consequences imperiling cell survival and overall health.
 |
|
Rocky
Tuan
|
Rocky S. Tuan received his Ph.D. in 1977 from the Rockefeller University in New York and was a postdoctoral fellow at Harvard Medical School in Boston, first in the Department of Orthopaedic Surgery at the Children’s Hospital and then in the Developmental Biology Laboratory at the Massachusetts General Hospital. In 1980, he joined the biology faculty at the University of Pennsylvania in Philadelphia, and in 1988 he became professor and director of Orthopaedic Research at the Thomas Jefferson University, Philadelphia, where, in 1997, he established the nation’s first Cell and Tissue Engineering Ph.D. program. In the fall of 2001, he joined the Intramural Research Program of NIAMS as chief of the newly created Cartilage Biology and Orthopaedics Branch.
Orthopaedic research is fundamentally a study of skeletal tissues and the biological activities that are important for their development, growth, function, and health. My lab is currently engaged in a variety of research projects, highlighted below, that focus on multiple aspects of skeletal and related biology. Our experimental approach integrates contemporary technologies of biochemistry, cell and molecular biology, embryology and development, and cellular imaging, as well as principles of bioengineering.
Cellular and Molecular Signaling during Cartilage Development. My lab has been studying the cellular and molecular mechanisms regulating chondrogenesis. A key working hypothesis is that cell-cell and cell-matrix interactions are important for chondrogenic differentiation. Our recent in vitro and in vivo findings show that the cell-cell adhesion molecule N-cadherin plays a critical role in the cellular condensation phase of chondrogenesis. Interestingly, we have also found that bioactive factors, such as BMP-2 and TGF-b1, which affect cartilage development, appear to act via the modulation of N-cadherin interaction with its cytosolic binding proteins, the catenins, which are also the target of the action of the Wnt family of signaling molecules. We are now investigating the exact, chondrogenically relevant signal transduction events regulated by the N-cadherin–catenin complex, as well as the signaling crosstalk with growth factors, such as those in the TGF-b superfamily (TGF-b1, BMP-2, and GDF-5), and Wnt family members and their cognate receptors, the Frizzled family.
We are also analyzing the possible involvement of another mode of cell-cell interaction, namely, gap junctional communication, specifically, the role of the gap junction protein connexin 43. We are currently studying limb mesenchymal chondrogenesis and the effect of growth factors of the TGF-b superfamily in transgenic mice harboring various constructs derived from the connexin 43 gene.
In terms of cell-matrix interactions, our investigations are focused on how fibronectin isoforms, produced sequentially in the developing limb bud, may provide important cell-matrix-mediated cues in the regulation of chondrogenesis and limb morphogenesis.
Molecular Biology of Axial Skeletal Patterning during Development. My lab has been examining the molecular aspects of somite formation—the first segmented patterning event in the early developing embryo—by focusing on the function of the Pax genes, a vertebrate gene family structurally related to the pair-rule genes of Drosophila, and of Paraxis, a member of the basic helix-loop-helix transcription factor gene family. We have used antisense technology to perturb the level of expression of these genes in the developing chick embryo and demonstrated the importance of Paraxis and Pax in somite epithelialization and differentiation-segmentation, respectively. We are currently carrying out gene-transfection experiments using the full-length Pax-1 cDNA to probe the downstream function as well as regulation of expression of the Pax-1 gene. Antisense experiments have also demonstrated that Paraxis expression is crucial for proper somitogenesis from the paraxial mesoderm of the segmental plate, most likely at the epithelialization step after somitomere formation. Currently we are using retroviral expression constructs of Paraxis to examine the effect of overexpression on somitogenesis. Finally, we have shown that misexpression of Pax-1 and/or Paraxis is functionally related to teratogen-induced somite dysmorphogenesis, based on our observations of chick embryos exposed to controlled heat shock, valproic acid, and carbon monoxide. This is an exciting finding because it represents the first correlation between somite teratogenesis, a segmentation gene, and a bHLH transcription factor. The potential implication in terms of axial skeletal birth defects, such as congenital scoliosis and Klippel-Feil syndrome, remains to be investigated.
Biology of Mesenchymal Progenitor Cells. On the basis of our previous findings on embryonic tissues, we hypothesize that multipotential mesenchymal progenitor cells reside as an endogenous cell population within a variety of mature, adult connective tissues, in addition to being a part of the marrow stroma. Our recent studies have revealed that mature human trabecular bone, derived from femoral head tissues obtained from total joint arthroplasty, indeed harbor cells with chondroprogenitor potential. This finding is consistent with the known ability of fractured bone to heal via the endochondral pathway, that is, the formation of cartilage, even in the absence of marrow cell proliferation. We are currently developing ways to optimize isolation and expansion of such cell lines, concurrent with clonal analysis and microarray gene expression profiling. With these approaches we hope to study the mechanisms responsible for chondrogenic differentiation in response to specific chondro-inductive environment.
Cartilage Tissue Engineering. Articular cartilage repair is an exciting challenge in musculoskeletal medicine. Using embryonic cell cultures, we have demonstrated proof of concept by using a solvent-leaching method to fabricate a porous, bio-resorbable, polylactide (PLA) scaffold and testing it as a delivery vehicle for peptide growth factors and as a cell-composite matrix for three-dimensional cartilage formation. These are exciting steps toward the development of new methodologies for in vitro three-dimensional cartilage tissue engineering that relies on bioresorbable polymers, such as PLA and polycaprolactone. We are pursuing three approaches to fabricating a three-dimensional scaffolding using human bone marrow stroma-derived mesenchymal stem cells: 1) press-coating to generate a thin hyaline cartilage surface coating for potential articular cartilage applications, 2) fabricating a PLA-alginate amalgam to optimize both mechanical and cellular requirements for chondrogenesis and cartilage formation, and 3) producing nanoscale fibers using electrospinning for cell seeding. We believe these new constructs may lead to the development of tissue-engineered osteochondral graft materials for clinical applications.
Bone-Implant Interaction. The long-term stability of an endoprosthetic device ultimately depends on the appropriate interaction between the implant and the host tissue and cells. My laboratory has been studying the cellular mechanisms involved in bone cell attachment to metallic orthopaedic surfaces. Our working hypothesis is that biomaterials that promote initial osteoblast adhesion will result in better osseointegration and contribute to a longer stability of the implant. In comparing several clinically used metallic alloys using a newly developed cell-adhesion assay, we have indeed observed a higher rate of osteoblast adhesion on Ti64, a titanium alloy commonly used in orthopaedic devices, particularly compared with standard tissue culture polystyrene. Our current research interests are in analyzing the sequence of cellular events responsible for this enhanced interaction between the bone cell and the underlying substratum, including adhesion, focal contact formation, matrix-integrin interactions, and signal transduction. We are studying these interactions via diverse techniques such as laser confocal microscopy, gene transfection, and receptor-ligand analysis. Thus far we have observed differences in the distribution of focal contacts and integrins, the cytoskeletal architecture, and cell spreading. Interestingly, osteoactive factors such as TGF-b1 and BMP-2 significantly enhance the initial cell adhesion event, suggesting that these factors may be candidates for enhanced osseointegration. Interestingly, our recent investigation into the mechanistic aspects shows that TGF-b1 treatment results in an immediate, transient intracellular Ca2+ flux, inhibitable by Ca2+ channel blockers, that is required in the adhesion enhancement. Preliminary analysis reveals altered phosphorylation of several intermediary signaling molecules. Our long-term goal is to develop a rational, cell-based set of parameters in evaluating bone implant interaction. We believe that this information will be invaluable for the practical design of orthopaedic implants.
Molecular Diagnosis and Experimental Therapeutic Treatment of Bone Infection. Bacterial infection is a major therapeutic challenge in orthopaedic surgery, more often than not complicated by the lack of rapid, sensitive, and reliable diagnosis. The conventional microbiological protocols are inherently slow and inaccurate. We have recently established a novel, rapid processing protocol for orthopaedically relevant tissue and fluid samples and its application in a polymerase chain reaction (PCR)–based detection of bacterial infections. The PCR protocol uses primers panspecific for the bacterial 16S rRNA gene and has the sensitivity to detect as few as 10 bacteria in the inoculum. Clinical studies are currently underway and the results so far support the applicability of the protocol. This method has already generated a great deal of attention in the orthopaedic community and has the potential to serve as a standard, adjunct algorithm for routine clinical diagnosis. Our present objective is to further develop the PCR-based protocol for speciation diagnosis, using gene sequence polymorphisms among different bacterial species.
Finally, despite the success of arthroplasty, periprosthetic infection remains a challenge. Our recent work has demonstrated, using animal models of osteomyelitis and open fracture, the efficacy of a PLA-based antibiotic delivery system for the prophylactic and therapeutic treatment of bone infection. We are currently investigating design modalities for surface modification of metallic implants for the purpose of controlled delivery of antimicrobial compounds to the bone-implant interface. Such materials would be ideal for efficient treatment of deep perioprosthetic infections.
 |
|
Ted
Usdin
|
Ted Usdin received M.D. and Ph.D. degrees from the Medical Scientist Training Program at Washington University in St. Louis in 1986. In 1990, after completing a residency in psychiatry at Stanford University, he joined the Laboratory of Cell Biology of NIMH. He is now a senior investigator in the Laboratory of Genetics, NIMH.
I am interested in the biological roles of neurotransmitters and neuromodulators. I am currently investigating the functions of TIP39 and the PTH2 receptor, a new neuropeptide and its receptor discovered in my laboratory. Following up on hints provided by our initial experimental observations, I am starting to investigate the potential roles of this peptide and receptor in pain modulation and endocrine function.
Several years ago, Tom Bonner of NIMH and I identified a receptor we named the PTH2 receptor (because it is the second known receptor activated by parathyroid hormone [PTH]) during a screen for new members of the secretin or Family B group of G-protein coupled receptors that are expressed in the CNS.
Éva Mezey of NINDS and I mapped the tissue and cellular distribution of the PTH2 receptor and found that it is expressed at greatest levels in the brain, where it is concentrated in several hypothalamic and limbic areas. However, we could find no evidence that PTH is synthesized in the brain. This launched me on a search for a different endogenous ligand for the PTH2 receptor. I used stimulation of cAMP production via the PTH2 receptor to screen tissue extracts for a selective receptor-stimulating activity. I found that a bovine hypothalamic extract stimulated the PTH2 receptor with much greater potency than the PTH1 receptor.
At the NCI Natural Products Support Group facility in Frederick, Tom McCloud and I extracted this activity from 50-pound batches of bovine hypothalamus. I purified the protein that produced the activity, determined its amino acid sequence, and used this information to have the peptide chemically synthesized and to determine its gene and cDNA sequences in human and mouse. This new peptide, tuberoinfund-ibular peptide of 39 residues (TIP39), is a distant relative of PTH and parathyroid hormone related–peptide. It potently simulates the PTH2 receptor and has no effect on the PTH1 receptor.
We have begun testing ideas about the function of TIP39 that are based on the anatomical distribution of the PTH2 receptor. For instance, the PTH2 receptor is expressed at particularly high levels in the outer layers of the dorsal horn of the spinal cord. This suggests that it may be involved in the modulation of pain perception. In collaboration with Hiroshi Ueda of Nagasaki University in Japan, we found that injecting TIP39 into a mouse paw elicits a withdrawal response, presumably by acting on PTH2 receptors in the peripheral terminals of primary afferent fibers. When we inject TIP39 intrathecally, mice scratch, bite, and lick their hindlimbs—a classic pain-avoidance response. Furthermore, intrathecal administration of an antibody that sequesters TIP39 decreases sensitivity to pain in several assays; injecting TIP39 has the opposite effect.
We are now mapping the anatomical distribution of TIP39. There are some TIP39-containing fibers in the spinal cord, consistent with its potential role as a supraspinal mediator of spinal pain processing. There are much denser networks of TIP39 fibers in other regions, and we are now beginning to test ideas about TIP39 function in these areas. There are only a small number of TIP39-containing neurons and they are largely concentrated in two brainstem nuclei. We want to find out what type of information reaches these cells and stimulates TIP39 release. We also want to find out whether TIP39 reaches peripheral PTH2 receptors, including those in pancreatic islets, thyroid parafollicular cells, and gastrointestinal peptidergic cells, and what functions it has in these regions.
Descriptive studies of PTH2 receptor distribution have led us to the discovery of a new neuropeptide and to the testing of specific hypotheses about its function. We expect that studying TIP39 will help us to learn more about physiological modulation, and possibly therapeutic intervention, in processes as diverse as pain perception and release of pituitary hormones.
 |
|
Ji
Ming Wang
|
Ji Ming Wang obtained his M.D. from the Graduate School of Shanghai Second Medical University, People’s Republic of China, in 1983. He then obtained his Ph.D. from the Lombardy Regional School for Professional Education in Pharmacology, Milan, Italy, in 1987. In 1990, he joined the Laboratory of Molecular Immunoregulation, Division of Basic Sciences (now the Center for Cancer Research), NCI-Frederick, where he is now a principal investigator.
My major research interests are the role of chemotactic factors (chemoattractants) and their cell receptors in health and disease conditions. Chemoattractants are a supergroup of molecules that induce directional migration (chemotaxis) of human cells. Chemotaxis is a crucial step for leukocyte accumulation at the sites of inflammation, bacterial infection, and immune responses where bacterial or tissue-produced chemoattractants are elevated. Chemoattractants are divided into "classical" and "chemokine" subgroups, and members of both subgroups use cell receptors with a seven-transmembrane structure. Some of the chemokine receptors act as co-receptors used by HIV for infection.
In the early 1990s, as a special volunteer and, later, as a contract scientist, I focused on the study of novel chemoattractants and receptors and the biological significance of these molecules. We were among the first to demonstrate the promiscuous binding nature of chemokines and identified novel receptors. We established with a mouse tumor model that chemokines are involved in the organ-preferential metastasis of malignant tumor cells, a principle that was confirmed by other investigators with human malignant tumor cells.
Since 1997, when I became a tenure-track investigator, my group has discovered that HIV-1 envelope proteins possess the capacity to inhibit the function of chemoattractant receptors on human mononuclear phagocytes. We further identified several peptide domains derived from HIV-1 envelope proteins that directly activate two receptors for the classical bacterial chemotactic peptides FPR and FPRL1. Activation of FPR and FPRL1 on human monocytes results in the deactivation of chemokine receptors, including a key HIV-1 co-receptor CCR5, and interferes with the ability of CCR5 to act as an HIV-1 co-receptor. These results suggest that FPR and FPRL1 on human cells may recognize HIV-1 envelope-derived peptides during the course of infection–perhaps reflecting a defensive host response to invading pathogens. However, prolonged activation of FPR and FPRL1 may lead to the suppression of cell response to stimulation by other chemoattractants, thereby toppling the balance of the immune system. On the bright side, peptides activating FPR and FPRL1—and thus inhibiting HIV-1 co-receptors—may provide a source of new anti–HIV-1 agents.
My group has also revealed an important role of the chemotactic peptide receptor FPRL1 in proinflammatory aspects of Alzheimer’s disease (AD). AD is characterized by the appearance of senile plaques in brain tissues, in association with progressive destruction of neurons and the development of dementia. The key causative factor of AD is a 42–amino acid b-amyloid peptide (Ab42). Production of Ab42 is increased in the context of pathologies such as genetic defects and brain injuries. Aggregated Ab42 forms the core of senile plaques and induces inflammatory responses, as shown by infiltration of the AD lesions by activated microglial cells, a cell type comparable to monocytes in the peripheral circulation.
We demonstrated that Ab42 uses FPRL1 as a functional receptor to induce migration and activation of monocytes and microglial cells. In addition, we detected a high level of expression of the FPRL1 gene in phagocytic cells in the lesions of the AD brain tissues. Furthermore, we found that FPRL1 is critical for cell uptake and intracellular accumulation of Ab42, culminating in the formation of amyloid aggregates and cell death. Interestingly, FPRL1 is also a functional receptor for a peptide fragment, Prp106-126, of the human prion protein, which causes kuru, a human version of bovine spongiform encephalopathy. Prp106-126 forms aggregates and promotes chemotaxis and production of neurotoxic mediators by mononuclear phagocytes. Identification of FPRL1 as a receptor for both Prp106-126 and Ab42 suggests similarity in the pathogenesis of prion diseases and AD.
In future studies, we will continue to focus on the elucidation of the molecular mechanisms underlying chemoattractant and receptor interactions. We are especially interested in using animal models to evaluate the contribution of these molecules to the progress of diseases. We hope the information obtained promotes understanding of the disease process and the design of novel therapeutic approaches.