
Steven
Holland
| T H E N I H C A T A L Y S T | S E P T E M B E R – O C T O B E R 2000 |
|
|
|
| P E O P L E |
RECENTLY TENURED
|
|
Steven
Holland
|
Steven Holland received his M.D. degree in 1983 from the Johns Hopkins University School of Medicine in Baltimore, where he was chief resident in internal medicine and a fellow in infectious disease before joining the Laboratory of Molecular Microbiology, NIAID, in 1989. In 1991, he joined the Laboratory of Host Defenses, NIAID, where he is now a senior clinical investigator.
My interests are broadly in the area of immunodeficiencies, especially as they affect phagocytes. I joined the laboratory as part of the research effort in chronic granulomatous disease (CGD), a pediatric immunodeficiency in which neutrophils fail to make superoxide and are therefore unable to produce hydrogen peroxide and bleach. The condition is characterized by severe recurrent bacterial and fungal infections, as well as severe granulomatous reactions that can obstruct internal organs and impair wound healing.
Early on, my colleagues and I used homologous recombination to create a mouse model of the most common autosomal recessive form of CGD. This model has furthered our understanding of granulomatous reactions. We have used it in gene therapy studies and are currently exploring some of the less anticipated ramifications of CGD. For instance, superoxide generated by the NADPH oxidase appears to be very important in the development of experimental allergic encephalomyelitis (EAE), ethanol-induced liver damage, and antigen-induced joint damage. We are using these animals, in conjunction with collaborators in the Laboratory Host Defenses and around the world to better dissect the critical roles that superoxide plays in signal transduction and immune modulation, as well as in host defense.
In caring for patients with CGD, I encountered a family with an apparent genetic susceptibility to mycobacterial infection. We treated these patients with interferon gamma, a critical downstream gene induced by IL-12, and had dramatic clinical results. Subsequently, Dave Frucht in the lab showed that this particular family has a problem affecting IL-12 production. This led us to develop a very successful clinical protocol using interferon gamma to treat patients with severe, refractory, nontuberculous mycobacterial infections.
In the course of our studies we identified several patients with genetic mycobacterial susceptibility who did not respond to interferon gamma therapy. Further study revealed associated mutations in interferon gamma receptor 1. With Susan Dorman in the lab, we then identified the first patients with mutations in interferon gamma receptor 2. This in turn has led us to collaborations with groups in Europe to identify the most common mutation in interferon gamma receptor 1, an autosomal dominant mutation at base 818.
We began these studies in mycobacterial disease in an effort to provide treatment for patients who were critically ill and had no further therapeutic options. As a result, we have been successful at identifying important pathways for future therapeutic exploration, as well as pathways that, when disrupted, lead to profound susceptibility to mycobacterial, fungal, and certain viral infections. It does not appear that general susceptibility to Mycobacterium tuberculosis is caused by mutations or polymorphisms in the same genes that cause susceptibility to nontuberculous infections. However, we have active protocols investigating the possibility that therapy using the interferon gamma and IL-12 pathways will be successful, especially in cases of severe multidrug resistant tuberculosis.
 |
|
Robert
Kreitman
|
Robert Kreitman received his M.D. in 1985 from Ohio State University in Columbus and completed clinical training in internal medicine at Duke University Medical Center, Durham, N.C., in 1988. During and after training in medical oncology at NIH , he worked in the Laboratory of Molecular Biology, NCI. He is now chief of the Clinical Immunotherapy Section in that lab, where he oversees translational and basic studies. He also serves as principal investigator for clinical trials conducted in the Clinical Center by the Medicine Branch of the NCI Division of Clinical Sciences.
I have been involved in the preclinical and clinical development of recombinant immunotoxins for the treatment of cancer and autoimmune disease. These agents contain an Fv domain, which binds to tumor-selective antigens, and a bacterial toxin (Pseudomonas exotoxin), which can kill a cell when only one or a few molecules reach the target cell’s cytoplasm. Recombinant immunotoxins are produced in large scale by plasmids in Escherichia coli. The resulting inclusion body protein is harvested and denatured, refolded in a redox buffer, and finally purified via ion-exchange and sizing chromatography.
In 1989, under the direction of Ira Pastan, I began testing the first recombinant immunotoxin made, anti-Tac(Fv)-PE40, which was engineered by Vijay Chaudhary. We tested this agent in freshly isolated peripheral blood malignant cells from patients with leukemia. The target cells included both B- and T-cell leukemias, Hodgkin’s disease (HD), and activated T-cells that mediate autoimmune disorders such as graft vs. host disease (GVHD). We chose freshly isolated cells because we realized that cancer cell lines are often unrealistic models for human leukemia—they display higher numbers of receptors and are more metabolically active and homogenous than malignant cells in patients. The binding portion of the agent is derived from Anti-Tac, the antibody to the interleukin-2 (IL-2) receptor alpha fragment (CD25) developed by Thomas Waldmann at NCI. We hypothesized that if fresh leukemic cells from patients could be killed by recombinant immunotoxin ex vivo, and comparable concentrations of immunotoxin could be reached in the blood of patients, then patients would also respond. We found anti-Tac(Fv)-PE40 and a slightly shorter derivative, anti-Tac(Fv)-PE38, nicknamed LMB-2, were, indeed, cytotoxic ex vivo against cells from a variety of leukemias, including adult T-cell leukemia (ATL), chronic lymphocytic leukemia (CLL), and hairy cell leukemia (HCL).
We next developed an animal model to evaluate the agent’s efficacy and safety in vivo and began a phase I trial of LMB-2 in 1996. I was principal investigator of the clinical protocol, in collaboration with co-investigators Wyndham Wilson of NCI’s Medicine Branch, Thomas Waldmann of NCI’s Metabolism Branch, and Ira Pastan of LMB. We found that LMB-2 was effective at the maximum tolerated dose, with major responses in ATL, CLL, cutaneous T-cell lymphoma, HD, and especially HCL, where one complete remission and three partial responses were observed in four patients who were resistant to chemotherapy.
Access to patient samples yielded new observations and conclusions about how immunotoxins poison cells and traffic to the cytoplasm. These studies also led to a better understanding of mechanisms of toxicity of immunotoxins in normal cells. We now plan to continue testing LMB-2 in patients with CD25+ malignancies and also in transplant patients, to prevent or treat GVHD.
In collaboration with Pastan and David FitzGerald of the LMB, we engineered the recombinant anti-CD22 immunotoxin, BL22, which improved targeting of B-cell malignancies. In preclinical studies, BL22 killed fresh human malignant B-cells ex vivo and also killed CD22+ tumors in mice. Since February 1999, I have treated 31 CD22+ CLL, HCL, or lymphoma patients with a total of 99 cycles of BL22. Both CLL and HCL patients have benefited clinically. BL22 is an excellent agent for either chemotherapy-refractory HCL or the poor-prognosis HCL variant, and many complete remissions have already been observed.
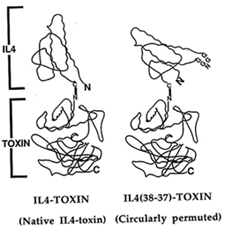 |
Several other agents produced in my lab are now being tested in clinical trials outside NIH. One of these is a circular IL-4 toxin, a single-chain recombinant toxin with the ligand-toxin junction at a location distinct from either the amino or carboxyl terminus of the ligand (see figure). This new ligand-toxin junction minimizes interference of the toxin with binding to the IL-4 receptor (IL-4R). Further preclinical and clinical development of this agent proceeded in collaboration with Pastan, Raj Puri of the FDA, and Robert Rand of the John Wayne Cancer Institute, Neurosurgery Department. in Los Angeles. The agent is currently being tested in patients with recurrent high-grade brain tumors, which are usually IL-4R+. The agent has already induced complete remissions and several other positive responses.
Finally, a fusion of granulocyte-macrophage colony stimulating factor with truncated diphtheria toxin (DTGM) was made in my lab and, in collaboration with Arthur Frankel of Wake Forest University in Winston-Salem, N.C., is now being tested in patients with recurrent acute myelogenous leukemia. Although test doses are currently very low, we have documented major positive responses.
Our goal is to continue developing recombinant immunotoxins as a therapeutic modality distinct from chemotherapy, surgery, and radiotherapy. These agents may be useful in patients failed by traditional treatments, but may also prove effective in synergistic combinations with chemotherapy, because their toxicities are quite different. My lab is now exploring new immunotoxins to target other antigens important in other types of hematologic malignancies.
Beyond these malignancies, I have recently begun trials of recombinant immunotoxins for solid tumors. Treating solid tumors with this approach is more challenging than hematologic malignancies, as the agents are more immunogenic in these patients and less able to penetrate solid tumors; nevertheless, we hope to be able to overcome these hurdles.
 |
|
Paul
Randazzo
|
Paul Randazzo earned his MD/PhD from Brown University in Providence, R.I. He received residency training in anatomic pathology and did postdoctoral work at the University of Pennsylvania in Philadelphia before joining NCI in 1990. He is a senior investigator in the Laboratory of Cellular Oncology.
I am interested in the regulation of the Arf family of monomeric GTP-binding proteins. The Arfs were first identified as cofactors for cholera-toxin–catalyzed ADP-ribosylation of the heterotrimeric G-protein, Gs. Subsequent studies have identified physiologic roles for Arf in membrane traffic and the actin cytoskeleton. Six mammalian Arfs have been identified, with multiple Arfs occurring within single cells.
Because a single Arf gene product can affect multiple membrane trafficking events, each Arf must be independently regulated. In addition, the Arfs affect constitutive as well as acutely regulated processes. The function of the Arfs, like other GTP-binding proteins, depends on the controlled binding and hydrolysis of GTP. Guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) control the cycle of GTP binding and hydrolysis and, as we and others are finding, contribute to Arf-specific and site-specific regulation. Some GEFs and GAPs are targets of signal transduction through cell surface receptors. Examination of the GEFs and GAPs has also been useful for understanding the function of Arf itself.
Recognizing that the GAPs might be site-specific regulators of specific Arfs and targets of signal transduction, we set out to purify a phosphoinositide-dependent Arf GAP. Through these efforts, we identified ASAP1 (Arf GAP with SH3, ANK repeat, and PH domains), a multidomain protein that contains phosphatidylinositol 4,5-bisphosphate–dependent GAP activity and binds the tyrosine kinase, Src. Subsequent studies found that ASAP1 is associated with and regulates focal adhesions. The Arf GAP activity is necessary but not sufficient to affect the focal adhesions, raising the possibility that ASAP1 is an Arf effector. Recent studies from other labs have found that ASAP1 can bind to paxillin and a GEF for Rho family proteins, PIX. Therefore, ASAP1 could be a key molecule in coordinating the activity of two families of GTP-binding proteins, the Arfs and Rhos, and, consequently, membrane traffic and actin cytoskeleton reorganization. One focus of the lab is to understand the molecular basis for ASAP1’s activity, including ASAP1’s role as a target for signal transduction through phosphoinositides and tyrosine kinases.
Based on sequence homology, a number of Arf GAPs related to ASAP1 have been identified, including PAP, ARAP1/2, ACAP1/2, and AGAP1/2. Like ASAP1, these proteins contain multiple domains that potentially interact with a number of signaling molecules. AGAPs contain a ras-like domain. They also have five PH domains and a Rho GAP domain. Preliminary studies indicate that all of these proteins affect cellular events that require coordination of membrane traffic and the actin cytoskeleton. By examining representative members of each family, we will test whether the Arf GAPs do provide Arf-specific and site-specific regulation and learn about the diversity of the signals targeting the Arf proteins. We also expect the studies will provide insights into the mechanisms regulating complex cellular events, like movement and phagocytosis, which involve membrane and actin remodeling.
 |
|
Thomas
Ried
|
Thomas Ried received his M.D. from the University of Heidelberg, Germany, in 1989 and did postdoctoral work in David Ward’s and Thomas Cremer’s laboratories (Yale University, New Haven, Conn., and Heidelberg, respectively). He joined NHGRI in 1994 and NCI in 1998. He is a senior investigator in the Genetics Department, Medicine Branch, DCS/NCI.
Comparative genomic hybridization (CGH) of more than 500 carcinomas in our laboratory has revealed that genomic imbalances, as a result of unbalanced chromosomal translocations or chromosomal gains and losses, are the premier cytogenetic event in solid tumors of epithelial origin.
Our CGH analyses have focused on colorectal adenomas, carcinomas, and metastases and ovarian carcinomas. We compare the cytogenetic profiles of these tissues with gene expression analyses to understand the functional consequences of chromosomal aneuploidy.
Our recent comparison of 25 solid-tumor cell lines using spectral karyotyping and CGH has provided compelling evidence that the vast majority of epithelial cancers are associated with aberrations in either the number or structure of chromosomes, resulting in genomic imbalances. The comparison of diploid, mismatch repair–deficient colorectal carcinomas with aneuploid carcinomas indicates that numerical chromosomal aberrations are 60 times as prevalent in the aneuploid tumors. Abnormalities of the centrosome are correlated with chromosomal aneuploidy both in human cancer cell lines and in animal cells deficient for cell-cycle regulator genes such as p53 and BRCA1. All these results taken together support the notion that chromosomal aneuploidy is a major theme in epithelial cancers.
In order to elucidate mechanisms leading to aneuploidy, to establish the functional relevance of chromosomal aneuploidy, and to identify whether aneuploidy is a cause or a consequence of genetic changes in solid tumors, we have focused on the following projects:
![]() Structural and functional analysis of abnormal centrosomes and their relationship
to chromosome segregation fidelity
Structural and functional analysis of abnormal centrosomes and their relationship
to chromosome segregation fidelity
![]() Live cell imaging of clonally derived cells after transfection with g-tubulin
green fluorescent protein (GFP) and CenPB-GFP vectors
Live cell imaging of clonally derived cells after transfection with g-tubulin
green fluorescent protein (GFP) and CenPB-GFP vectors
![]() Sequential inhibition of p53 and Rb function with E6 and E7 genes from human
papilloma virus 16 and subsequent analyses of chromosomal instability
Sequential inhibition of p53 and Rb function with E6 and E7 genes from human
papilloma virus 16 and subsequent analyses of chromosomal instability
![]() Microcell-mediated chromosome transfer and in vitro induction of chromosomal
aneuploidy in cells derived from normal colorectal epithelium and adenomas,
followed by cytogenetic analysis and assays for cellular immortalization and
transformation
Microcell-mediated chromosome transfer and in vitro induction of chromosomal
aneuploidy in cells derived from normal colorectal epithelium and adenomas,
followed by cytogenetic analysis and assays for cellular immortalization and
transformation
![]() Comparison of cells carrying chromosomal aneuploidies with their wild-type parental
cells using cDNA microarrays and 2-D gel protein electrophoresis
Comparison of cells carrying chromosomal aneuploidies with their wild-type parental
cells using cDNA microarrays and 2-D gel protein electrophoresis
Efforts to define the sequence of genetic aberrations during human carcinogenesis increasingly depend on animal models, as does the understanding of the biology of tumor progression and the development of test systems for novel therapeutics. Murine models of human carcinogenesis are widely used to delineate genetic mechanisms that determine tumor initiation and progression, and improved methods for genetic manipulation open new avenues to study biological pathways of tumorigenesis. For this reason, we have worked hard to develop molecular cytogenetic tools to analyze chromosomal aberrations in mouse models of human cancer. Karyotype analyses of chemically induced plasmacytomas in mice, in lymphomas from ATM- or Ku80-deficient animals, and in mice transgenic for c-myc have provided evidence for the conservation of mechanisms leading to chromosomal aberrations across species boundaries. We have also been able to validate mouse models of human cancers by comparing the pattern of cytogenetic abnormalities with orthologous maps of human chromosomes.
Finally, we are interested in applying the knowledge of specific chromosomal aberrations to the diagnosis of cancer in cytological preparations. For instance, more than 90 percent of cervical carcinomas carry extra copies of chromosome 3, and virtually all diploid breast cancers show a gain of chromosome 1q. Also, gains of chromosome 3q precede copy number increases of chromosomes 5p and 1 and loss of chromosome 2 during the genesis of cervical carcinomas. It is therefore helpful to use these recurring and specific chromosomal aberrations to complement and enhance the cytomorphological diagnosis of human cancers and their precursor lesions. This can be achieved using cytogenetics with fluorescently tagged DNA probes that recognize specific chromosomal target regions directly in interphase cells. We have focused on three applications:
![]() Identification of the progressive potential of cervical intraepithelial neoplasia
based on the detection of extra copies of chromosomes 3 and 5 in thin-prep PAP
smears
Identification of the progressive potential of cervical intraepithelial neoplasia
based on the detection of extra copies of chromosomes 3 and 5 in thin-prep PAP
smears
![]() Visualization of specific chromosomal aneuploidies in cytokeratin-positive epithelial
cells isolated from the peripheral blood of breast cancer patients
Visualization of specific chromosomal aneuploidies in cytokeratin-positive epithelial
cells isolated from the peripheral blood of breast cancer patients
![]() Diagnosis and prognostication of suspicious breast lesions following the detection
of chromosomal aneuploidies in fine needle aspirates
Diagnosis and prognostication of suspicious breast lesions following the detection
of chromosomal aneuploidies in fine needle aspirates
![]()