
A
view of cell death from the Apoptosis
Interest Group web site
| T H E N I H C A T A L Y S T | N O V E M B E R – D E C E M B E R 1999 |
|
|
|
Research FestivitiesCELL SOLILOQUY:TO DIE OR NOT TO DIE |
by Cynthia Delgado |
|
|
A
view of cell death from the Apoptosis
Interest Group web site
|
When confronted by damaged DNA, cells can either set about repairing themselves or commit suicide through programmed cell death. How the cell "decides" which path it will follow was explored during one of the NIH Research Festival’s mini-symposiums—"Survival or Death: DNA Repair or Apoptosis." Chaired by Vilhelm Bohr, chief of the NIA Laboratory of Molecular Genetics, and Curtis Harris, chief of the NCI Laboratory of Human Carcinogenesis, the symposium focused on the pathways and players in these life-and-death cellular processes, as elucidated in ongoing studies in NIH labs.
One of the key players in DNA repair is DNA polymerase beta (b pol), which is instrumental in each of two basic excision repair (BER) pathways—single nucleotide and long-patch—and whose absence leads to "drastic" consequences, according to Rob Sobol, a research fellow in the NIEHS Laboratory of Structural Biology. Sobol presented findings from b pol knockout studies, conducted with NIEHS Deputy Director Samuel Wilson, chief of the DNA Repair and Nucleic Acid Enzymology Workgroup, that showed that "b pol is required for protection against the induction of cytotoxic hypersensitivity, apoptosis, and chromosomal breakage caused by exposure to methylating agents." Sobol also reviewed the findings of studies conducted by Wilson in collaboration with NIA’s Bohr, in which b pol was shown to be the DNA polymerase of choice involved in the long-patch BER pathway in mammalian cells (1–3). Moreover, one of b pol’s major enzymatic roles, the removal of the dRP group, was found to be the overall rate-limiting step of BER (4). He and other investigators, Sobol added, have recently demonstrated that mouse fibroblasts from b pol knockouts are highly sensitive to induction of apoptosis and chromosomal breakage by methylating agents, further emphasizing b pol’s protective role (5).
 |
 |
 |
 |
 |
 |
|
Samuel
Wilson
|
Rob
Sobol
|
Xin
Wang
|
Albert
Fornace
|
Vilhelm
Bohr
|
Curtis
Harris
|
Bohr elaborated on the use of Cockayne syndrome (CS)—a premature aging syndrome characterized by cell sensitivity to ultraviolet (UV) light, a deficiency or delay in DNA and RNA synthesis after irradiation, and a defect in repair of active genes—as a model to better understand DNA repair dynamics, such as transcription-coupled DNA repair (6). He also recapped mounting evidence that CS involves not only a repair defect but also a transcription defect (7).
Another major player in DNA repair is the well-known tumor suppressor p53. While noting p53’s more popularly studied activities in cell-cycle delay and apoptosis, Albert Fornace, of NCI’s Division of Basic Science, emphasized its equally valid "protective role in modulating cellular levels of DNA repair."
"The bottom line," Fornace said, "is that transcription-coupled repair does not appear to be perturbed appreciably by the loss of p53, one of the major mediators of cytotoxicity, after UV radiation." He added, however, that "global repair is attenuated" in cells lacking p53. His lab recently published in Nature Genetics the "striking find" that disruption of the p53-effector gene, GADD45a, results in genomic instability in a manner reminiscent of that seen in p53-null mice. Observations included centrosome amplification, chromosome aberrations, aneuploidy, mitosis abnormalities, and increased radiation-induced carcinogenesis in GADD45a-null cells (8). Interestingly, he said, disruption of GADD45a also resulted in reduced DNA-repair capacity. Compared with wild-type cells, Gadd45a-null cells demonstrated a significant reduction in both unscheduled DNA synthesis and global excision repair. As in p53-null cells, transcription-coupled repair was not reduced, whereas repair of UV-type damage to the nontranscribed strand or to bulk DNA (global repair) was reduced. These results, Fornace said, indicate that some of the features of p53 that contribute to genomic stability and efficient DNA repair are mediated by the product of the GADD45a gene.
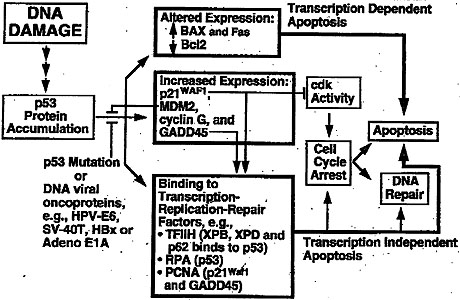 |
|
A simplistic model of p53’s place on a DNA damage-response pathway (absent a cellular context and the influence of the cellular microenvironment. From X. Wang and C. Harris. "p53 tumor-suppressor gene: clues to molecular carcinogenesis." J. Cell. Physiol. 173: 247—255 (1997). |
p53, said Xin Wang, of the NCI Laboratory of Human Carcinogenesis, stands "at the crossroads of life and death," a pivotal player in crucial biochemical pathways such as transcription, DNA repair, cell-cycle control, and apoptosis. Wang pointed out that many human malignancies have a p53 mutation and that other investigators have shown that the loss of p53 can result in genomic instability, increased chromosomal rearrangements, and mutation. He and his co-workers, therefore, reason they may find "clues to molecular carcinogenesis" by analyzing the interactive roles of p53.
Human diseases that have a genomic instability or cancer predisposition—such as xeroderma pigmentosum, ataxia telangectasia, Werner’s syndrome, and Bloom’s syndrome—may assist them in unraveling the complex network of cross-talks mediated through p53, he said. For example, Wang speculates that in the p53-mediated apoptotic pathway, p53 may induce apoptosis by binding to and inhibiting the helicase activity of certain transcription factors.
This helicase-binding
motif is also found on the DNA helicase BLM, the gene product of the Bloom’s
syndrome gene, BS. Wang said he expects these studies to enhance understanding
of human cancer and ultimately to facilitate the development of effective new
anticancer strategies. ![]()
References
1. G.L. Dianov, R. Prasad, S.H. Wilson, and V.A. Bohr. "Role of DNA polymerase b in the excision step of long patch mammalian base excision repair." J. Biol. Chem. 274: 13741—13743 (1999).
2. R. Prasad, G.L. Dianov, V.A. Bohr, and S.H. Wilson. "FEN1 Stimulation of DNA polymerase b mediates an excision step in mammalian long patch base excision repair." J. Biol. Chem. (in press).
3. J.K. Horton, R. Prasad, E.W. Hou, and S.H. Wilson. "Protection against methylation-induced cytotoxicity by DNA polymerase b-dependent long patch base excision repair." J. Biol. Chem. (in press).
4. D.K.Srivastaua, B.J. Vande Berg, R. Prasad, J.T. Molina, W.A.Beard, A.E. Tomkinson, and S.H. Wilson. "Mammalian abasic site base excision repair." J. Biol. Chem. 273: 21203—21209 (1998).
5. K. Ochs, R.W. Sobol, S.H. Wilson, and B. Kaina. "Cells deficient in DNA polymerase beta are hypersensitive to alkylating agent-induced apoptosis and chromosomal breakage." Cancer Res. 59: 1544—1551 (1999).
6. R.M. Brosh, A.S. Balajee, R. Selzer, M. Sunesen, L.P. De Santis, and V.A. Bohr. "The ATPase domain, but not the acidic region of Cockayne syndrome group B gene product is essential for DNA repair." Mol. Biol. Cell (in press).
7. M.C. Hollander, M.S. Sheikh, D.V. Bulavin, K. Lundgren, L. Augeri-Henmueller, R. Shehee, T.A.Molinaro, K.E. Kim, E. Tolosa, J.D. Ashwell, M.P Rosenberg, Q. Zhan, P.M. Fernandes-Salguero, W.F. Morgan, C.X. Deng, A.J. Fornace, Jr. "Genomic instability in GADD45a-deficient mice." Nat. Genet. 23: 176—184 (1999).
8. E.S. Spillare, A. Roble, X.W. Wang, J. Shen, C. Yu, G.D. Schellenberg, and C.C. Harris, "p53-mediated apoptosis is attenuated in Werner syndrome cells." Genes Dev. 13: 1355—1360 (1999).